Durable disease control in a radiation-induced high-grade glioma harboring NF1 and PTPN11 co-mutations
Mohamed Sherief, Maria Fatteh, Jaime Wehr, Matthias Holdhoff, Charles G Eberhart, Valsamo Anagnostou, Karisa C Schreck

TL;DR
A patient with a rare brain tumor caused by past radiation was treated successfully for 20 months using a targeted therapy based on genetic testing.
Contribution
Demonstrates the clinical benefit of MEK inhibition in a radiation-induced glioma with specific co-mutations.
Findings
The patient had a high-grade glioma with NF1 and PTPN11 mutations after prior radiation.
Treatment with trametinib led to 20 months of disease control.
Genomic profiling identified actionable mutations in the MAPK pathway.
Abstract
Radiation-induced gliomas (RIGs) are rare and aggressive secondary brain tumors arising years after cranial irradiation. Their management remains challenging due to prior radiation exposure, which limits additional radiation, and a lack of effective chemotherapies. Recent studies have revealed distinct molecular profiles in RIGs with unclear clinical implications. This study presents the case of an individual who developed a high-grade glioma three decades after curative craniospinal radiation for medulloblastoma. He was treated with repeat radiation and temozolomide chemotherapy but developed recurrence with disseminated leptomeningeal disease thereafter. Molecular profiling of the tumor revealed a loss-of-function NF1 mutation and a gain-of-function PTPN11 mutation, two convergent alterations in the MAPK pathway. Based on these findings, the patient was treated with a MEK inhibitor,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
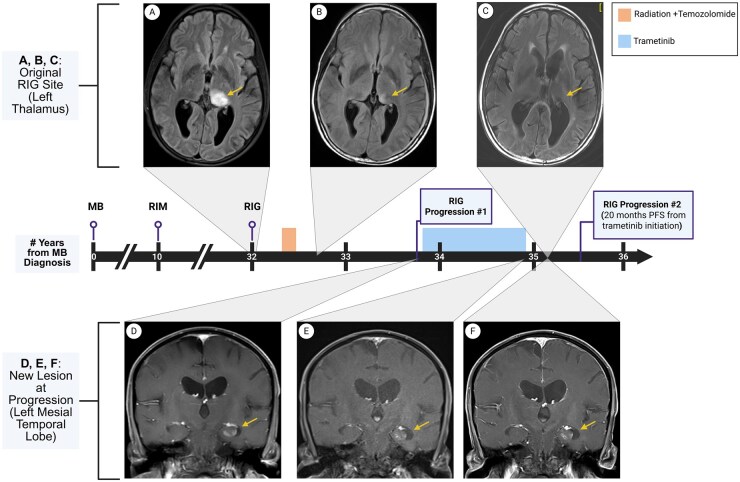 Figure 1
Figure 1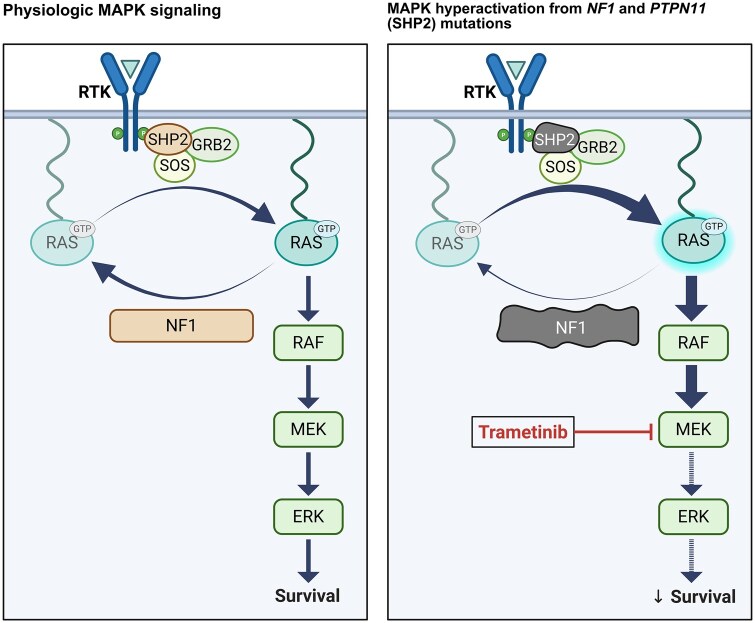 Figure 2
Figure 2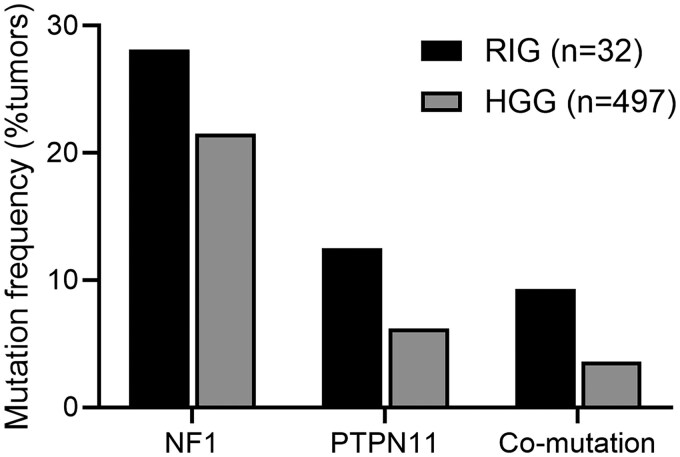 Figure 3
Figure 3- —Department of Defense10.13039/100000005
- —Doris Duke Career Development
- —Jane and Robert Gore Family Fund for Cancer Research
- —Johns Hopkins Department of Neurology
- —Sidney Kimmel Comprehensive Cancer Center10.13039/100011566
- —United States National Institutes of Health ITCR
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBrain Metastases and Treatment · Glioma Diagnosis and Treatment · Melanoma and MAPK Pathways
Introduction
Radiation-induced gliomas (RIGs) are a rare yet serious late complication of cranial irradiation, often arising years or decades after the initial exposure. These tumors typically present with high-grade histopathology (eg glioblastoma [GBM]) and carry a dismal prognosis, with a reported median overall survival averaging 7 to 11 months.1 In the absence of specific guidelines for RIGs, current management strategies largely reflect those of de novo high-grade glioma (HGG), typically involving re-irradiation and chemotherapy. Upon the inevitable disease progression, options become significantly limited, as additional radiation is often unfeasible due to prior exposure and surgical re-resection is challenging. Salvage chemotherapy provides only marginal benefits, highlighting the need for novel therapeutic strategies in these patients, particularly those guided by tumor molecular profiling.
In recent years, comprehensive genomic characterization of RIGs has indicated that RIGs harbor distinct genetic alterations compared to their de novo counterparts.2–4 These include recurrent structural alterations such as PDGFRA and CDK4 amplifications and CDKN2A/B homozygous deletions, as well as mutations in the mitogen-activated protein kinase (MAPK) pathway genes including NF1.2–4 Classical alterations associated with spontaneous HGGs such as IDH1/2, PTEN, or TERT are infrequent in RIGs. Despite advances in comprehensive genomic profiling, due to the rarity of this tumor, no clinical trials have specifically evaluated targeted therapies for RIGs, and most patients continue to rely on conventional treatments that yield suboptimal outcomes.
Here, we present the case of a patient with a high-grade RIG harboring a loss-of-function (LOF) mutation in NF1 and a gain-of-function (GOF) mutation in PTPN11, both key regulators of the MAPK pathway. Given this molecular profile, the patient was treated with a MEK inhibitor, trametinib, at time of disease recurrence, attaining disease control for 20 months. This case highlights the importance of genomic profiling in RIGs and the potential utility of molecularly targeted approaches for patients with RIGs.
Case Report
The patient is a 45-year-old male with a history of medulloblastoma, of unknown molecular subtype, diagnosed at age 10. He underwent gross total resection, followed by craniospinal irradiation (CSI; 3600 cGy with a posterior fossa boost to 5600 cGy), and adjuvant chemotherapy with vincristine, lomustine, and cisplatin. At age 20, he was diagnosed with a right frontal meningioma, presumed to be radiation-induced, for which he underwent curative surgical resection.
At age 42, 32 years after his initial medulloblastoma diagnosis, the patient developed progressive leftward gaze dysfunction, headaches, and worsening left-sided motor deficits. Brain MRI revealed a left thalamic FLAIR -hyperintense lesion that increased in size over 2 months, prompting further evaluation with a stereotactic biopsy (Figure 1A). Histopathologic evaluation showed features consistent with high-grade astrocytic glioma, IDH-wildtype. To further classify the tumor, molecular profiling was performed. FISH analysis revealed a 1p deletion with intact 19q. Next‑generation sequencing (NGS) using the Caris MI Profile comprehensive assay, which incorporates whole-exome and whole-transcriptome sequencing, detected a pathogenic NF1 p.R681* (variant allele frequency [VAF], 62%) and PTPN11 p.T507K (VAF, 32%) mutation, along with an unmethylated MGMT promoter. The tumor mutation burden was low (5 mutations/Mb) and PD-L1 immunohistochemistry was negative. Given the patient’s history of CSI, the histopathology and molecular profiling supported an integrated histopathologic diagnosis of high-grade RIG, consistent with previous studies reporting similar molecular alterations in RIGs.2^,^3 The tumor was classified as high-grade with a Ki-67 proliferation index exceeding 20%.
Timeline of diagnosis, treatment, and radiographic response to trametinib. (A) T1-weighted post-gadolinium MRI of the original lesion in the left thalamus at initial presentation, (B) 3 months post-chemoradiation showing treatment response, and (C) 15 months after the initiation of trametinib showing stability. (D) A new lesion in the left mesial temporal lobe was identified 15 months post-chemoradiation, showing disease progression (Progression #1). Treatment with trametinib was initiated, and MRI (E) after 12 months of treatment and (F) 3 months after trametinib discontinuation confirmed stable disease, prior to a second progression that occurred 20 months after trametinib initiation (Progression #2). Abbreviations: MB, medulloblastoma; RIM, radiation-induced meningioma; RIG, radiation-induced glioma; PFS, progression-free survival; MRI, magnetic resonance imaging. Created in BioRender. Schreck, K. (2026) https://BioRender.com/57ob9ut.
The patient completed a 6-week course of proton radiotherapy to a total dose of 5460 cGyE administered concurrently with temozolomide. Proton therapy was selected given the patient’s prior cranial irradiation, with the goal of minimizing cumulative radiation exposure to surrounding normal brain tissue while adhering to dose constraints for adjacent critical structures, including the brainstem. Given the tumor’s unmethylated MGMT promoter status that typically confers resistance to temozolomide, adjuvant temozolomide was deferred, and he transitioned to active surveillance. Initial follow-up MRI demonstrated early tumor response with continued mass shrinkage (Figure 1B). However, at 15 months post-chemoradiation, surveillance MRI revealed disease progression with the emergence of two new lesions: an 8 mm enhancing nodule in the left mesial temporal lobe and a 3-5 mm enhancing nodule along the ependymal surface of the left occipital horn, with no progression at the primary site (Figure 1C and D). These were concerning for leptomeningeal dissemination given their proximity to the lateral ventricle.
Given the presence of NF1 and PTPN11 mutations, both of which drive constitutive MAPK activation and were deemed oncogenic by the Johns Hopkins Molecular Tumor Board, targeted therapy was recommended. Additionally, given the occurrence of two radiation-associated neoplasms, comprehensive germline testing was recommended; however, the patient and family declined. The patient did not meet eligibility criteria for a clinical trial and therefore began off-label monotherapy with the MEK inhibitor trametinib at the FDA-approved dose of 2 mg daily. After two months of treatment, he developed drug-induced pneumonitis, prompting a temporary treatment hold for 1 month. Following resolution of treatment-related toxicity, he resumed trametinib at a reduced dose of 1 mg daily and continued therapy for a total of 12 months before discontinuation due to declining functional status and the duration of treatment. MRI at the time of discontinuation demonstrated stable disease in the left mesial temporal lobe lesion and complete disappearance of the left occipital horn ependymal nodule, with no evidence of new enhancement or progression (Figure 1E). Follow-up MRI 3 months post-discontinuation confirmed continued stable disease (Figure 1F). Twenty months after initiating trametinib and seven months after drug discontinuation, the patient developed two new ependymal nodules consistent with disease progression.
Discussion
MAPK Pathway Activation in Gliomas is Clinically Actionable
The MAPK pathway plays a central role in gliomagenesis and progression and can be activated through upstream RTK alterations, loss of negative regulators such as NF1, or activating mutations in effectors like BRAF. Given frequent dysregulation, the MAPK pathway has emerged as a therapeutic target in gliomas. MEK inhibitors such as trametinib and selumetinib have demonstrated notable clinical benefit in a select low-grade glioma (LGG) subtypes, particularly pediatric gliomas with BRAF V600E mutations or arising in the context of an NF1 predisposition syndrome.5^,^6 There are no published clinical data on the use of MEK inhibitors in RIGs, a tumor type that, based on recent molecular profiling studies, appears molecularly distinct from de novo HGGs, with frequent MAPK pathway alterations, and follows an aggressive clinical course.2–4 Here, we report the case of a patient with a high-grade RIG harboring concurrent NF1 and PTPN11 mutations who attained durable disease control following treatment with trametinib.
NF1 and PTPN11 Co-Mutation May Confer MAPK Pathway Dependence and Sensitivity to MEK Inhibition in HGGs
The rationale for using MEK inhibition in this case stems from the biological consequences of the NF1 p.R681* truncating mutation identified in the tumor. NF1 encodes the RAS-GTPase activating protein neurofibromin, a negative regulator of RAS, and its loss leads to constitutive RAS activation and sustained MAPK signaling (Figure 2). NF1 mutations are well-documented in both RIGs and de novo HGGs, where they cluster with the mesenchymal subtype of GBM.2 Preclinical studies in NF1-deficient GBM models have demonstrated sensitivity to MEK inhibition through suppression of MAPK activity and reduced tumor proliferation.7^,^8 Clinically, however, only a few reports have documented modest responses to trametinib monotherapy in NF1- mutated HGGs, particularly GBM. For instance, a subprotocol of the NCI-MATCH trial reported that among five GBM patients with deleterious NF1 alterations, two showed clinical benefit, with the longest reported PFS reaching 9.2 months.9
Schematic of MAPK pathway activation and therapeutic inhibition in NF1/PTPN11-mutated glioma. Under physiological conditions, MAPK signaling is tightly regulated. NF1 negatively regulates RAS activity, and SHP2 (encoded by PTPN11) remains autoinhibited until activated by upstream RTKs (left panel). In tumors with NF1 loss and activating PTPN11 mutations, RAS becomes constitutively active, leading to sustained MAPK signaling; MEK inhibition by trametinib reduces ERK activation and impairs downstream survival signaling (right panel). Abbreviations: RTK, receptor tyrosine kinase; MAPK, mitogen-activated protein kinase; NF1, neurofibromin 1; PTPN11, protein tyrosine phosphatase non-receptor type 11; SHP2, Src homology region 2-containing protein tyrosine phosphatase-2. Created in BioRender. Schreck, K. (2026) https://BioRender.com/57ob9ut.
While NF1 LOF provides a compelling rationale for targeting MAPK signaling, the presence of a concurrent activating PTPN11 mutation further reinforces MAPK pathway dependence and introduces additional considerations for pathway regulation and therapeutic responsiveness. A recent multi‑omic study identified PTPN11 and its protein phosphorylation status as a central node of oncogenic signaling in HGGs, and clinical evidence further suggests that mutations in PTPN11 are associated with poor prognosis in GBM.10 PTPN11 encodes SHP2, a protein tyrosine phosphatase that normally remains in an autoinhibited conformation. Upon stimulation by phosphorylated RTKs, SHP2 transitions to an active conformation, regulating multiple downstream signaling pathways, including the MAPK pathway (Figure 2).11 GOF mutations, such as the PTPN11 p.T507K that lies in the phosphatase domain, can disrupt SHP2’s autoinhibition, and lead to constitutive phosphatase activity, resulting in sustained RAS pathway signaling, and additional input into the MAPK cascade, amplified by NF1 loss.
While this convergence on MAPK signaling may enhance sensitivity to MEK inhibition, it also raises questions regarding potential mechanisms of resistance. Preclinical studies, including those in NF1-deficient cancers and BRAF V600E-mutant HGGs, have shown that MEK inhibitor resistance can emerge through loss of negative feedback leading to activation of upstream RTKs, a process in which SHP2 upregulation plays a central role.12^,^13 These observations prompted ongoing preclinical and clinical investigations into dual MEK and SHP2 inhibition as a strategy to overcome adaptive resistance to MAPK pathway inhibition, though no SHP2 inhibitors are yet FDA approved. It remains unknown whether combined therapy would prevent adaptive resistance in cancers with combined NF1 and PTPN11 alterations.
NF1 and PTPN11 Co-Mutations are Enriched in RIGs but also Present in a Subset of de novo HGGs
Although RIG is a rare subset of HGG, a similar NF1/PTPN11 co-mutation pattern has been previously reported in some of the few available genomic studies of this entity. For example, Whitehouse et al. described a patient with a RIG harboring both NF1 and PTPN11 mutations, in which the corresponding patient-derived xenograft exhibited high levels of phosphorylated ERK, consistent with MAPK pathway activation.14 This observation was further supported by one of the largest genomic analyses of RIG to date, in which DeSisto et al. identified NF1 mutations in 9 of 32 patients (28.1%), PTPN11 mutations in 4 (12.5%), and NF1/PTPN11 co-mutations in 3 patients (9.3%).2 Notably, in the same study, trametinib emerged as one of the most active agents in an in vitro drug screen of RIG-derived cell lines, although drug responses were not stratified by mutational profile.
Whether this therapeutic vulnerability to MEK inhibition, observed in this case of NF1/PTPN11 co-mutated RIG, represents a molecular synergy unique to RIGs or extends to a broader subset of de novo HGGs, a more common and molecularly heterogeneous tumor type, remains to be determined. To further explore the occurrence of NF1/PTPN11 co-mutations, we queried a publicly available dataset of GBMs from the MSKCC glioma cohort via cBioPortal (http://www.cbioportal.org). Among 497 GBM tumors, NF1 mutations were identified in 107 (21.5%), PTPN11 mutations in 31 (6.2%), and co-mutations in 18 tumors (3.6%). While the co-mutation frequency was lower than that observed in DeSisto RIG cohort (9.3%), its presence suggests that this alteration pattern is not unique to RIGs (Figure 3). Notably, this co-occurrence pattern aligns with findings from a recent analysis of NF1-mutant GBMs, which reported that tumors with NF1 mutations were significantly more likely to harbor co-mutations in PTPN11 compared to NF1 wild-type tumors. Together, these findings raise the possibility that a subset of de novo HGGs may share similar MAPK pathway dependencies—and potentially similar sensitivity to MEK inhibition—as observed in this case.
Frequency of NF1 and PTPN11 mutations in RIG and sporadic HGG. Percentage of tumors with NF1 (n=9 out of 32, 28.1%), PTPN11 (n=4 out of 32, 12.5%), or NF1 + PTPN11 co-mutations (n=3 out of 32, 9.3%) in RIGs (n=32) are shown alongside frequencies of tumors with NF1 (n=107 out of 497, 21.5%), PTPN11 (n=31 out of 497, 6.2%), or NF1 + PTPN11 co-mutations (n=18 out of 497, 3.6%) in sporadic HGG (n=497). RIG data were derived from the DeSisto et al cohort; HGG data were obtained from the MSKCC glioma dataset via cBioPortal. Abbreviations: RIG, radiation-induced glioma; HGG, high-grade glioma.
Potential for MEK Inhibition as a Radiosensitization Strategy in NF1/PTPN11-Mutated RIG
Building on recent preclinical work by Ioannou et al., which demonstrated that MEK inhibition enhances radiotherapy efficacy in NF1-deficient GBM models, one of which also harbors a PTPN11 alteration, and earlier studies showing that SHP2 inhibition increases glioma radiosensitivity, our observation of trametinib’s benefit in an NF1/PTPN11-mutated RIG suggests this synergy could extend to RIGs.15 Further work will be necessary to understand whether combining MEK inhibitors with radiotherapy could enhance tumor control or allow for radiation dose de-escalation, given the prior radiation exposure in RIGs patients.
This case highlights the value of comprehensive molecular profiling in RIGs and presents clinical evidence for MEK inhibitor activity in a MAPK pathway-addicted RIG. While single-patient outcomes must be interpreted with caution, the observed 20-month disease control period in this case supports the therapeutic potential of targeting MAPK signaling in NF1/PTPN11 co-mutated gliomas. Future studies are needed to validate these findings and explore rational treatment strategies, such as dual MEK/SHP2 inhibition and radiosensitization approaches, in both de novo and secondary HGGs harboring similar molecular features.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Onishi S , Yamasaki F, Amatya VJ, et al Characteristics and therapeutic strategies of radiation-induced glioma: case series and comprehensive literature review. J Neurooncol. 2022;159:531-538. 10.1007/s 11060-022-04090-935922583 · doi ↗ · pubmed ↗
- 2De Sisto J , Lucas JT Jr., Xu K, et al Comprehensive molecular characterization of pediatric radiation-induced high-grade glioma. Nat Commun. 2021;12:5531. 10.1038/s 41467-021-25709-x 34545084 PMC 8452624 · doi ↗ · pubmed ↗
- 3Deng MY , Sturm D, Pfaff E, et al Radiation-induced gliomas represent H 3-/IDH-wild type pediatric gliomas with recurrent PDGFRA amplification and loss of CDKN 2A/B. Nat Commun. 2021;12:5530. 10.1038/s 41467-021-25708-y 34545083 PMC 8452680 · doi ↗ · pubmed ↗
- 4López GY , Van Ziffle J, Onodera C, et al The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol. 2019;137:139-150. 10.1007/s 00401-018-1906-z 30196423 PMC 6589431 · doi ↗ · pubmed ↗
- 5Bouffet E , Geoerger B, Moertel C, et al Efficacy and Safety of Trametinib Monotherapy or in Combination With Dabrafenib in Pediatric BRAF V 600-Mutant Low-Grade Glioma. J Clin Oncol. 2023;41:664-674. 10.1200/JCO.22.0100036375115 PMC 9870224 · doi ↗ · pubmed ↗
- 6Fangusaro J , Onar-Thomas A, Young Poussaint T, et al Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncology. 2019;20:1011-1022. 10.1016/S 1470-2045(19)30277-3 · doi ↗
- 7See WL , Tan IL, Mukherjee J, Nicolaides T, Pieper RO. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res. 2012;72:3350-9. 10.1158/0008-5472.CAN-12-033422573716 PMC 4128256 · doi ↗ · pubmed ↗
- 8Schreck KC , Allen AN, Wang J, Pratilas CA. Combination MEK and m TOR inhibitor therapy is active in models of glioblastoma. Neurooncol Adv. 2020;2:vdaa 138. 10.1093/noajnl/vdaa 13833235998 PMC 7668446 · doi ↗ · pubmed ↗