Total Synthesis of (+)-Malbrancheamide B Employing a Bioinspired Strategy
M. Aurelia Bosi, Radek Pohl, Ullrich Jahn

TL;DR
This paper describes a new method for the total synthesis of the complex natural compound (+)-malbrancheamide B using a bioinspired approach.
Contribution
The paper introduces a novel bioinspired synthetic strategy for assembling the complex structure of (+)-malbrancheamide B.
Findings
A bioinspired diketopiperazine assembly was used as a key step in the synthesis.
A stereoselective oxidative radical cyclization provided a three-dimensional bridged diketopiperazine precursor.
An intramolecular Friedel–Crafts cyclization formed the hexacyclic skeleton of the target compound.
Abstract
Prenylated indole alkaloids are a class of secondary metabolites isolated from various terrestrial and marine fungi. The common complex structure, featuring a diazabicyclo[2.2.2]octane scaffold fused to a tetrahydrocarbazole unit, and the numerous biological activities make these compounds attractive synthetic targets. In the present work an asymmetric total synthesis of (+)-malbrancheamide B is reported. Key steps are bioinspired diketopiperazine (DKP) assembly, a stereoselective oxidative radical cyclization, which gives access to the three-dimensional bridged diketopiperazine precursor and an intramolecular Friedel–Crafts cyclization providing the hexacyclic skeleton of malbrancheamide B.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
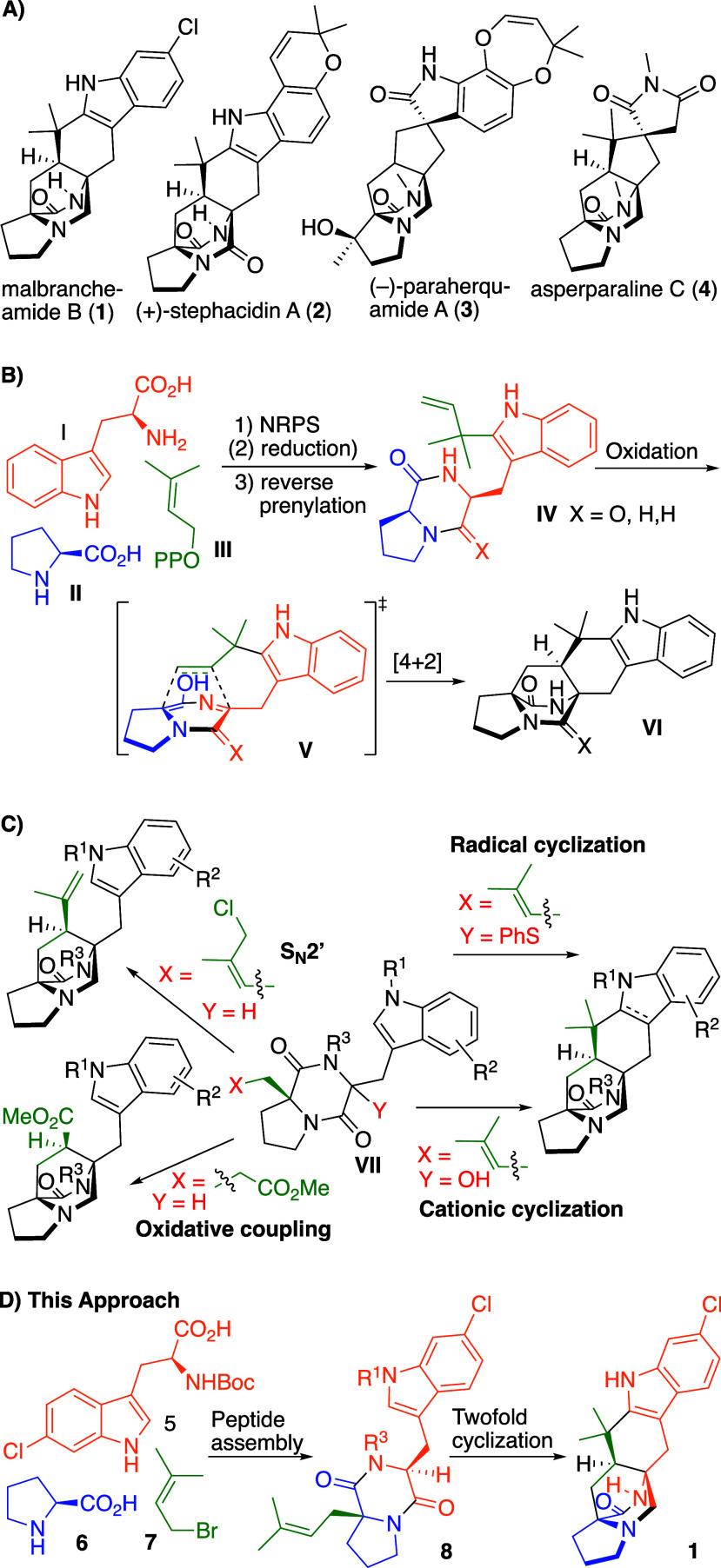 Figure 1
Figure 1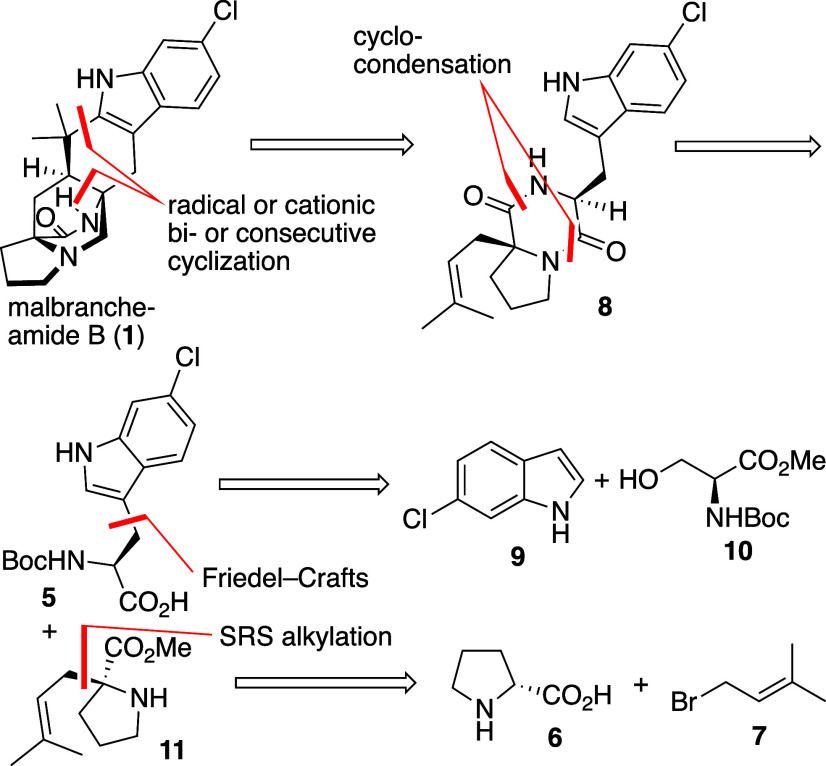 Figure 2
Figure 2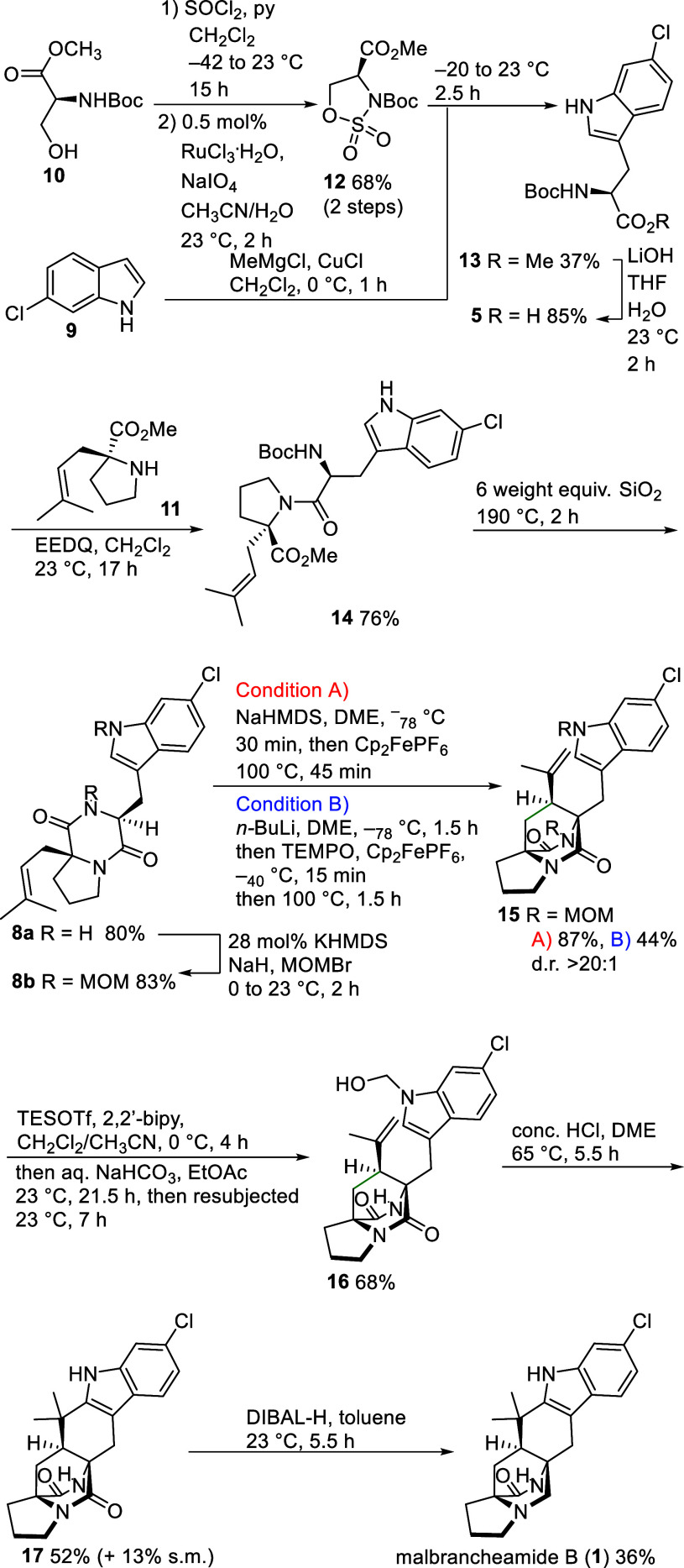 Figure 3
Figure 3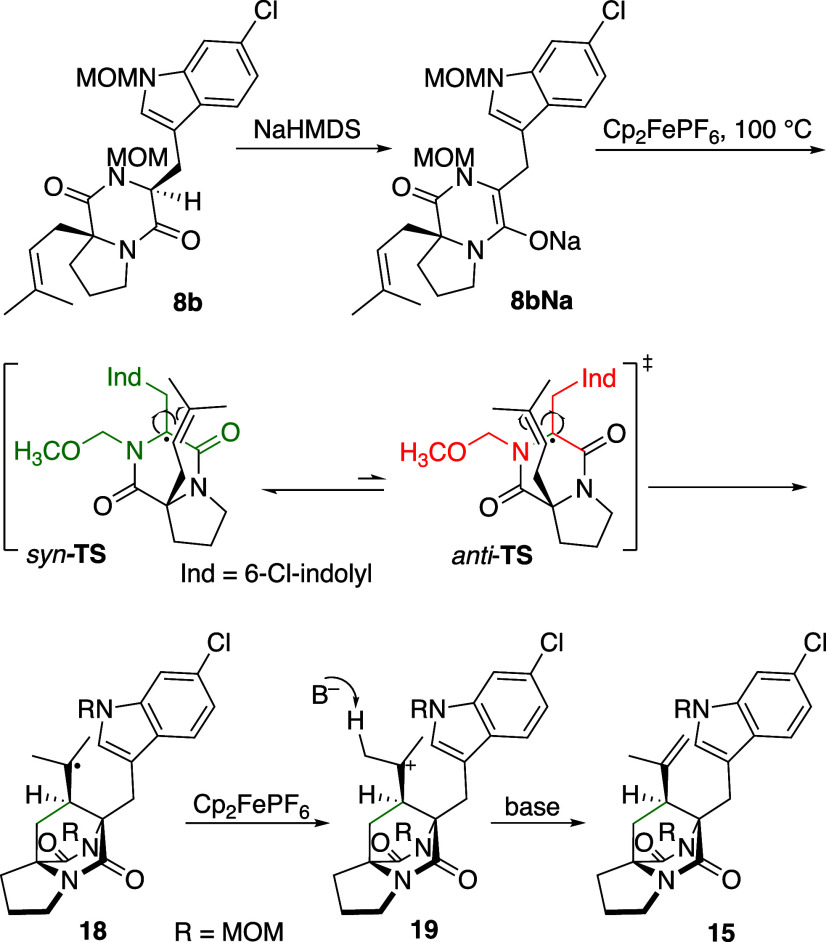 Figure 4
Figure 4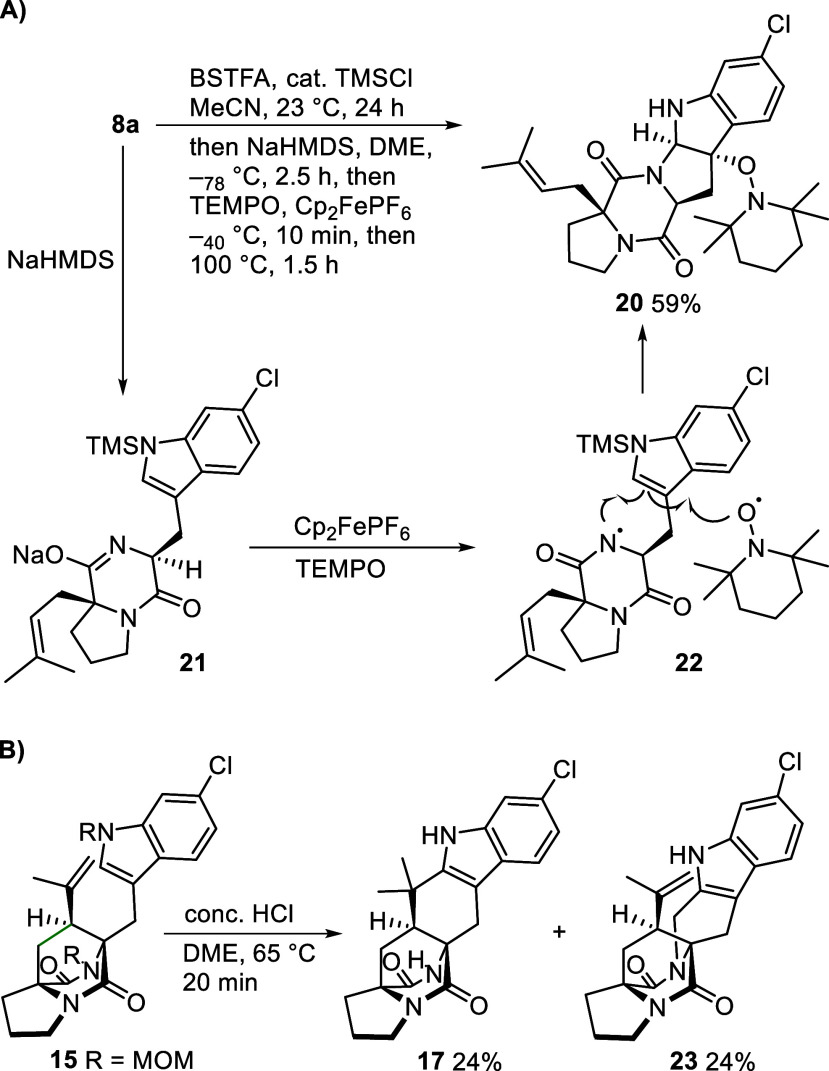 Figure 5
Figure 5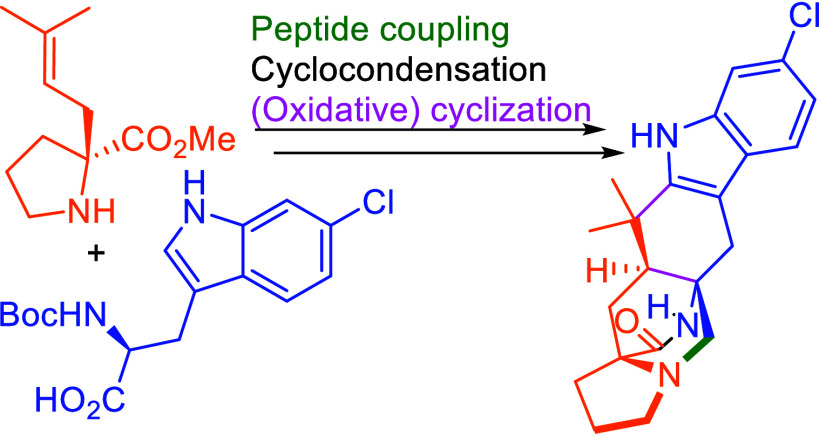 Figure 6
Figure 6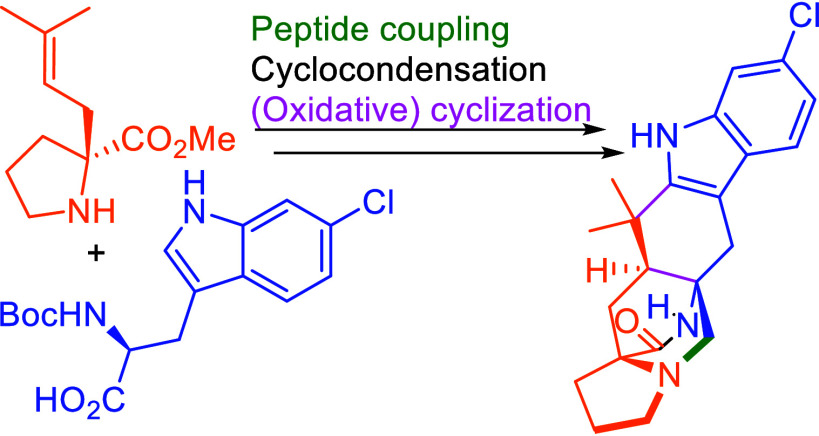 Figure 7
Figure 7- —Univerzita Karlova v Praze10.13039/100007397
- —European Regional Development Fund10.13039/501100008530
- —?stav organick? chemie a biochemie Akademie ved Cesk? republiky10.13039/501100010099
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMarine Sponges and Natural Products · Synthetic Organic Chemistry Methods · Chemical synthesis and alkaloids
Bridged (di)ketopiperazine alkaloids, ?,? such as malbrancheamide B (1),? stephacidin A (2),? paraherquamide A (3)? or asperparaline C (4),? are a part of the broad class of terpenylated indole alkaloids predominately isolated from a variety of terrestrial and marine fungi (FigureA). ?,? These natural products exhibit a diketopiperazine scaffold and a prenyl bridge connecting its 3- and 6-positions, thus forming a unique diazabicyclo[2.2.2]octane system, which is linked in fused or spiro mode to indole, oxoindole, or indoxyl rings. ?,? Other prominent members are the brevianamides,? marcfortines,? notoamides,? or taichunamides? (not shown). In the malbrancheamides, marcfortines, asperparalines, and paraherquamides one of the two amide carbonyl groups is reduced to a methylene. The alkaloids display a broad range of biological activities, being anthelmintic, insecticidal, cytotoxic, or antibacterial, ?,? whereas malbrancheamide B is a calmodulin inhibitor and vasorelaxant. ?,?
Several studies have focused on the biosynthesis of prenylated indole alkaloids (FigureB). A general pathway involving nonribosomal dipeptide synthesis (NRPS) from tryptophan I and proline II, reverse prenylation with III in 2-position of the tryptophan unit, and an oxidative intramolecular Diels–Alder reaction of DKP IV to construct the bridged core structure VI was substantiated ?,?−? ? ? ? and is supported by ab initio calculations.? This occurs for most natural products with syn diastereoselectivity of the hydrogen atom and the bridging secondary amide, although a few alkaloids have anti configuration. ?,?
Their complex structures and numerous biological activities make these natural products attractive targets in total synthesis. ?,?,?,? Five general strategies have been applied for total syntheses of bridged diketopiperazine alkaloids VI. The Williams, ?,?,? Scheerer? and recently the Banwell? groups developed biomimetic intramolecular Diels–Alder cycloadditions (cf. FigureB). This approach leads, however, mostly to racemic alkaloids and syn/anti mixtures since the stereochemical information in the DKP precursor IV is destroyed during generation of azadiene V, unless residing stereocenters exert a directing effect. ?,? All other approaches are intermediate-based and relied on prefunctionalized precursors VII (FigureC). ?,?,? Williams and colleagues used chloroprenyl precursors in a stereoselective S_N_2′ cyclization to construct the bridge for the synthesis of several bridged DKP alkaloids. ?,? An oxidative ester enolate-indole coupling was used by Baran et al.? in the total synthesis of 2. Simpkins and co-workers employed in contrast bridge-head functionalized diketopiperazines and applied them in radical cascade cyclizations to hexacycles with an indoline unit, which was transformed to stephacidin A (2),? or cationic cascade cyclizations providing brevianamide B and ent-malbrancheamide B.?
Based on our experience in the synthesis of diketopiperazines, ?,? we hypothesized that a bioinspired direct approach for the total synthesis of malbrancheamide B (1) may be accomplished. This is enabled by a strategy relying on peptide assembly using chlorotryptophan 5, proline 6 and prenyl bromide 7 to achieve a direct access to prenylated diketopiperazine precursor 8 (FigureD), which was envisaged to provide the natural product 1 by an oxidative bicyclization or consecutive cyclization reactions. Here we report the large potential of this approach, which results in a short total synthesis of the natural product.
Reducing this bioinspired approach to practice requires the following retrosynthesis (Scheme). The removal of the oxo group to accomplish the total synthesis of malbrancheamide B (1) is proposed to take place as the last step. Further disconnection at the quaternary center next to the indole unit and at the diketopiperazine bridge by a radical bicyclization or consecutive radical and cationic cyclizations calls for diketopiperazine 8. A retro-cyclocondensation leads to the corresponding amino acid derivatives 5 and 11. Chlorotryptophan 5 can be traced to chloroindole 9 and serine ester 10; this mimics the biosynthesis of tryptophan. 2-Prenylproline ester 11 will be approached from d-proline 6 and prenyl bromide 7 using Seebach’s “self-regeneration of stereocenters (SRS)” methodology.?
The synthesis commenced with commercial N-Boc-l-serine methyl ester 10, which was transformed into sulfamidate 12 by treatment with thionyl chloride in the presence of pyridine, followed by oxidation with sodium periodate and a catalytic amount of ruthenium(III) chloride in analogy to Zervosen et al. (Scheme).? Chloroindole 9 was metalated at nitrogen according to the method developed by Piersanti and co-workers? and the resulting indolylcopper intermediate was treated with 12 leading to chlorotryptophan methyl ester 13. The moderate yield is in line with those observed by Piersanti et al.;? however, this approach is the most straightforward and general to obtain substituted tryptophans. Other pursued approaches starting from tryptophan methyl ester are longer and not higher yielding (see the Supporting Information (SI)). Saponification of the methyl ester with lithium hydroxide provided the N-Boc amino acid 5. The second amino acid building block, α-prenyl proline methyl ester 11, was prepared from d-proline using Seebach’s SRS strategy in good yield, ?,? The subsequent peptide coupling was conducted using N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) as coupling agent in the absence of base, to prevent epimerization at the tryptophan unit, affording dipeptide 14 as a single diastereomer in good yield.
The thermal cyclization of dipeptide 14 at high temperature on silica gel under strictly inert conditions provided diketopiperazine 8a in very good yield as a single diastereomer. This method is practical since absorbing the dipeptide at silica gel prevents substrate diffusion, side reactions and allows a very simple purification.? Protection of both the amide and the indole nitrogen atoms in 8a was necessary at this point (vide infra), the methoxymethyl (MOM) group was chosen because of its stability under basic conditions required in the next step. In the event, twofold MOM protection of DKP 8a was not trivial; standard conditions with NaH? gave only 27% yield (not shown). However, introducing a substoichiometric amount of a soluble base such as potassium bis(trimethylsilyl)amide (KHMDS) in tetrahydrofuran (THF) facilitated the protection significantly, providing a very good yield of 8b. Acyl or carbamate protecting groups were not applicable since a Chan–Lam-type rearrangement leading to ring contraction is known to take place when deprotonating acyl-protected DKPs at the α-position.? The application of a protected precursor for the cyclization is mandatory, since otherwise other pathways prevail (vide infra, Scheme).
The crucial oxidative bridge-forming cyclization was performed by two methods. Reasoning that the DKP enolate precursor may be thermally rather robust, under condition A) a ferrocenium ion-mediated oxidative cyclization was leveraged to form the diazabicyclo[2.2.2]octane ring system. Diketopiperazine 8b was deprotonated at low temperature and subsequently heated to 100 °C and ferrocenium hexafluorophosphate was added to trigger the cyclization. This proved indeed highly practical and provided bridged DKP 15 essentially as a single *syn-*diastereomer. In contrast a 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO)-intercepted cyclization (condition B)), which was inspired by a previous total synthesis of the bridged DKP unit in asperparaline C (4),? was much less efficient. Single-electron oxidation of the lithium enolate of 8b at low temperature in the presence of TEMPO results in the formation of an extremely labile adduct, which decays on slow warming. To facilitate the cyclization, the reaction mixture was transferred to a preheated bath triggering the radical cyclization and hydrogen atom transfer. The isopropenyl-substituted bridged DKP 15 was isolated in moderate 44% yield, but also as a single *syn-*diastereomer.
The mechanism and diastereoselectivity of the oxidative cyclizations of 8b can be explained as follows (Scheme). Deprotonation of diketopiperazine 8b generates the corresponding enolate 8bNa. Subsequent single-electron oxidation leads to selective radical generation at the α-position of the tryptophan unit. The subsequent 6-exo cyclization proceeds essentially exclusively via *syn-*transition state syn-TS because the orientation of the cyclizing prenyl unit toward the less demanding carbonyl group is strongly preferred over the alternative arrangement in anti-TS where interactions with the methylene groups of the MOM and tryptophan units may hamper reaching the cyclization transition state. The cyclized tertiary alkyl radical 18 is subsequently oxidized to the corresponding carbocation 19.
At this stage an intramolecular Friedel–Crafts-type cyclization was envisaged to take place providing the complete malbrancheamide skeleton. However, the basic conditions of the reaction favored deprotonation to isopropenyl diazabicyclo[2.2.2]octandione 15. Under the previously established cyclization conditions? TEMPO couples with the initial DKP radical at low temperature. Heating the labile adduct also triggers the cyclization via syn- TS; however, this proved to be less effective (not shown).
To complete the total synthesis, compound 15 was initially subjected to aqueous HCl to initiate the crucial cationic cyclization and concomitant MOM deprotection (Scheme). However, this provided an unexpected outcome (vide infra, Scheme). Therefore, initial MOM deprotection was mandatory, which was not trivial. After much experimentation (see the SI), reaction with triethylsilyl triflate using 2,2′-bipyridine as a formaldehyde scavenger proved optimal.? The resulting mixture was subjected to basic conditions leading to slow, but complete lactam deprotection after resubjection of the reaction mixture to aqueous NaHCO_3_ solution, but the hydroxymethyl group at the indole unit was resistant to removal. Bridged diketopiperazine 16 subsequently cyclized in the presence of hydrochloric acid giving oxomalbrancheamide B 17 in 52% yield; 13% of starting material (s.m.) was recovered under the conditions. Gratifyingly, the remaining hydroxymethyl group was also removed under the cyclization conditions. At this stage the total synthesis converged with Simpkins’s^22^ and Scheerer’s? total syntheses after only nine steps. A final diisobutylaluminum hydride (DIBAL-H) reduction according to the literature? provided malbrancheamide B (1) after 10 steps in 1.2% overall yield.
Several investigations support the pursued strategy. In parallel to the successful strategy an in situ-protection/deprotection methodology was tested (SchemeA).? In short, diketopiperazine 8a was treated with an excess bis(trimethylsilyl) trifluoroacetimidate (BSTFA) in the presence of catalytic chlorotrimethylsilane. After evaporation, the resulting protected diketopiperazine intermediate was deprotonated with sodium bis(trimethylsilyl)amide (NaHMDS) and subjected to cyclization condition B) (cf. Scheme). However, pentacyclic product 20 was isolated in 59% yield instead of the expected bridge formation.
This suggests that the diketopiperazine NH in 8a is not protected under the reaction conditions. Therefore, on deprotonation with NaHMDS amidate 21 was generated, which is oxidized to amidyl radical 22. A rare radical 5-endo cyclization and coupling with TEMPO provides the product. Compound 20 is related to DKP-bound pyrroloindoline alkaloids, another attractive class of diverse biologically active alkaloids.? The presence of the alkoxyamine group might allow further diversification toward the synthesis of these target molecules.
A direct Friedel–Crafts-type cyclization of bis(MOM)-protected bridged diketopiperazine 15 was also entertained (SchemeB). However, an inseparable 1:1 mixture of the desired cyclization product 17 and indole aminomethylation product 23 was isolated. This shows that MOM protection has to precede cyclization to avoid the formation of a reactive N-acyliminium ion, which competes for the indole with the intramolecular Friedel–Crafts-type cyclization.
In conclusion, an asymmetric bioinspired total synthesis of 1 has been achieved over 10 steps in 1.2% overall yield. Key steps are a biomimetic dipeptide assembly, diketopiperazine formation, and a diastereoselective oxidative radical cyclization which enables the construction of the bridged diketopiperazine motif by reverting the polarity at the α-position of the amide and forming a new C–C bond. The full skeleton was assembled by an intramolecular Friedel–Crafts-type cyclization. This blueprint will serve to approach other relevant bridged indolodiketopiperazine alkaloids and their biological investigation.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Finefield J. M.Frisvad J. C.Sherman D. H.Williams R. M.Fungal origins of the bicyclo[2.2.2]diazaoctane ring system of prenylated indole alkaloids J. Nat. Prod.20127581283310.1021/np 200954 v 22502590 PMC 3485739 · doi ↗ · pubmed ↗
- 2Du L.Dian L.Newmister S. A.Xia Y.Luo G.Sherman D. H.Li S.The mutually inspiring biological and chemical synthesis of fungal bicyclo[2.2.2]diazaoctane indole alkaloids Chem. Rev.20251251718180410.1021/acs.chemrev.4c 0025039927617 PMC 11936112 · doi ↗ · pubmed ↗
- 3a Figueroa M.González M. D. C.Mata R.Malbrancheamide B, a novel compound from the fungus Malbranchea aurantiaca Nat. Prod. Res.20082270971410.1080/1478641080201236118569711 · doi ↗ · pubmed ↗
- 4Qian-Cutrone J.Huang S.Shu Y.-Z.Vyas D.Fairchild C.Menendez A.Krampitz K.Dalterio R.Klohr S. E Gao Q.Stephacidin A and B: two structurally novel, selective inhibitors of the testosterone-dependent prostate LN Ca P cells J. Am. Chem. Soc.2002124145561455710.1021/ja 028538 n 12465964 · doi ↗ · pubmed ↗
- 5Yamazaki M.Okuyama E.Kobayashi M.Inoue H.The structure of paraherquamide, a toxic metabolite from Penicillium paraherquei Tetrahedron Lett.19812213513610.1016/0040-4039(81)80168-2 · doi ↗
- 6Hayashi H.Nishimoto Y.Akiyama K.Nozaki H.New paralytic alkaloids, asperparalines A, B and C, from Aspergillus japonicus JV-23Biosci. Biotechnol. Biochem.20006411111510.1271/bbb.64.11110705455 · doi ↗ · pubmed ↗
- 7Birch A. J.Wright J. J.The brevianamides: a new class of fungal alkaloid J. Chem. Soc. D 1969644 b 64510.1039/c 2969000644 b · doi ↗
- 8a Polonsky J.Merrien M.-A.Prange T.Pascard C.Moreau S.Isolation and structure (X-ray analysis) of marcfortine A, a new alkaloid from Penicillium roqueforti J. Chem. Soc., Chem. Commun.198060160210.1039/C 39800000601 · doi ↗