An Unexpected Isomerization for the Total Synthesis of Daphnepapytone A
Ilja Lubins, Bernhard Breit

TL;DR
The paper describes a high-yielding synthesis of daphnepapytone A using unexpected isomerization to improve yield.
Contribution
A novel isomerization strategy is introduced to convert unwanted byproducts into the desired compound during synthesis.
Findings
A trisubstituted chiral cyclopentenone was efficiently synthesized via reductive cleavage.
A serendipitous aldehyde epimerization improved the yield by converting unwanted exo-epimer into the endo-enantiomer.
The final natural product was obtained in fewer steps using a second Pauson–Khand reaction.
Abstract
We report a high-yielding total synthesis of sesquiterpenoid daphnepapytone A from (S)-glycidol. The groundwork for the synthesis is laid via reductive cleavage of a bicyclic Pauson–Khand-derived cyclopentenone ether, giving efficient access to a trisubstituted chiral cyclopentenone which is difficult to obtain by other means. To introduce the cyclobutane motif, the enone was irradiated in the presence of allene gas, yielding the unwanted exo-cyclobutane at worst and 1:1 mixtures of the endo- and exo-product at best. A serendipitous epimerization of an aldehyde in the following steps converted the unwanted exo-epimer into the endoenantiomer, which significantly improved the final yield. The final cage was connected with a second Pauson–Khand reaction, requiring only one more step to yield the natural product.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
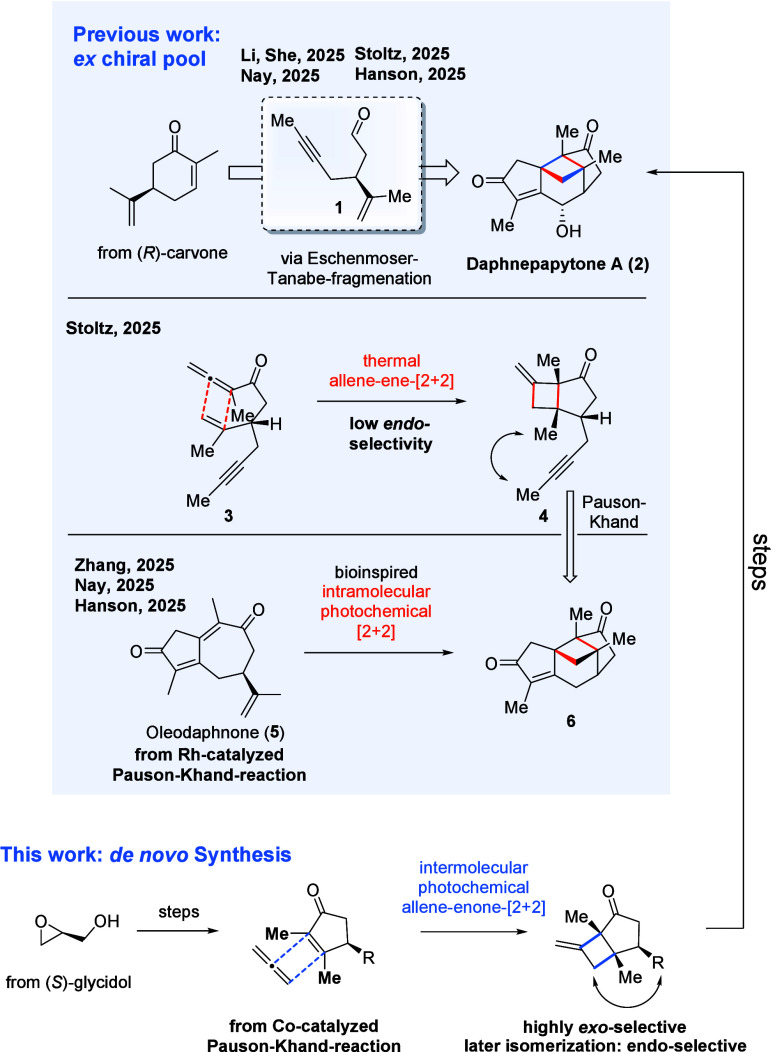 Figure 1
Figure 1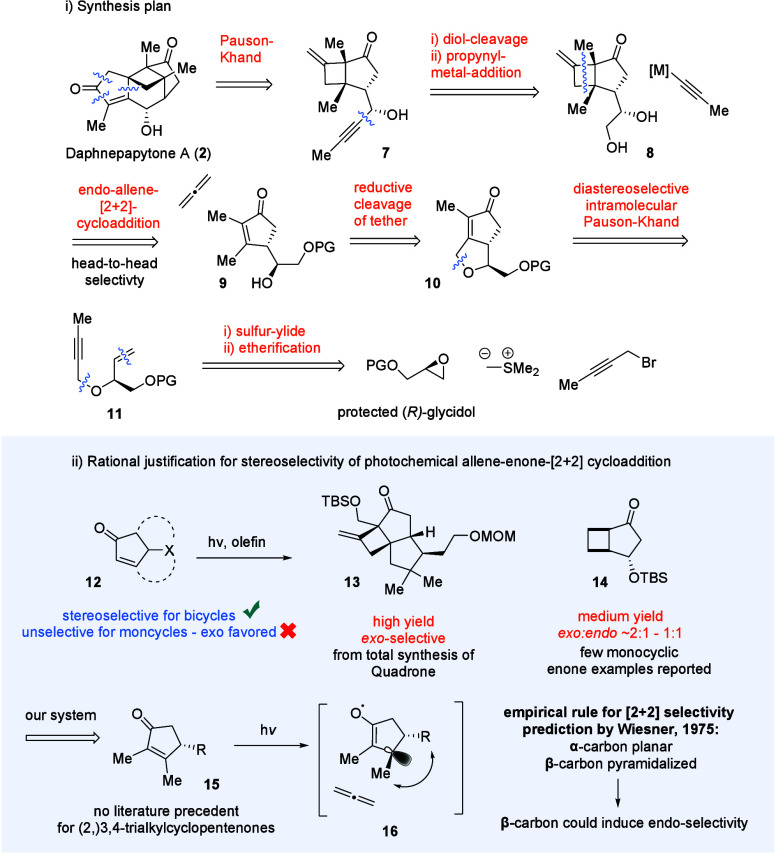 Figure 2
Figure 2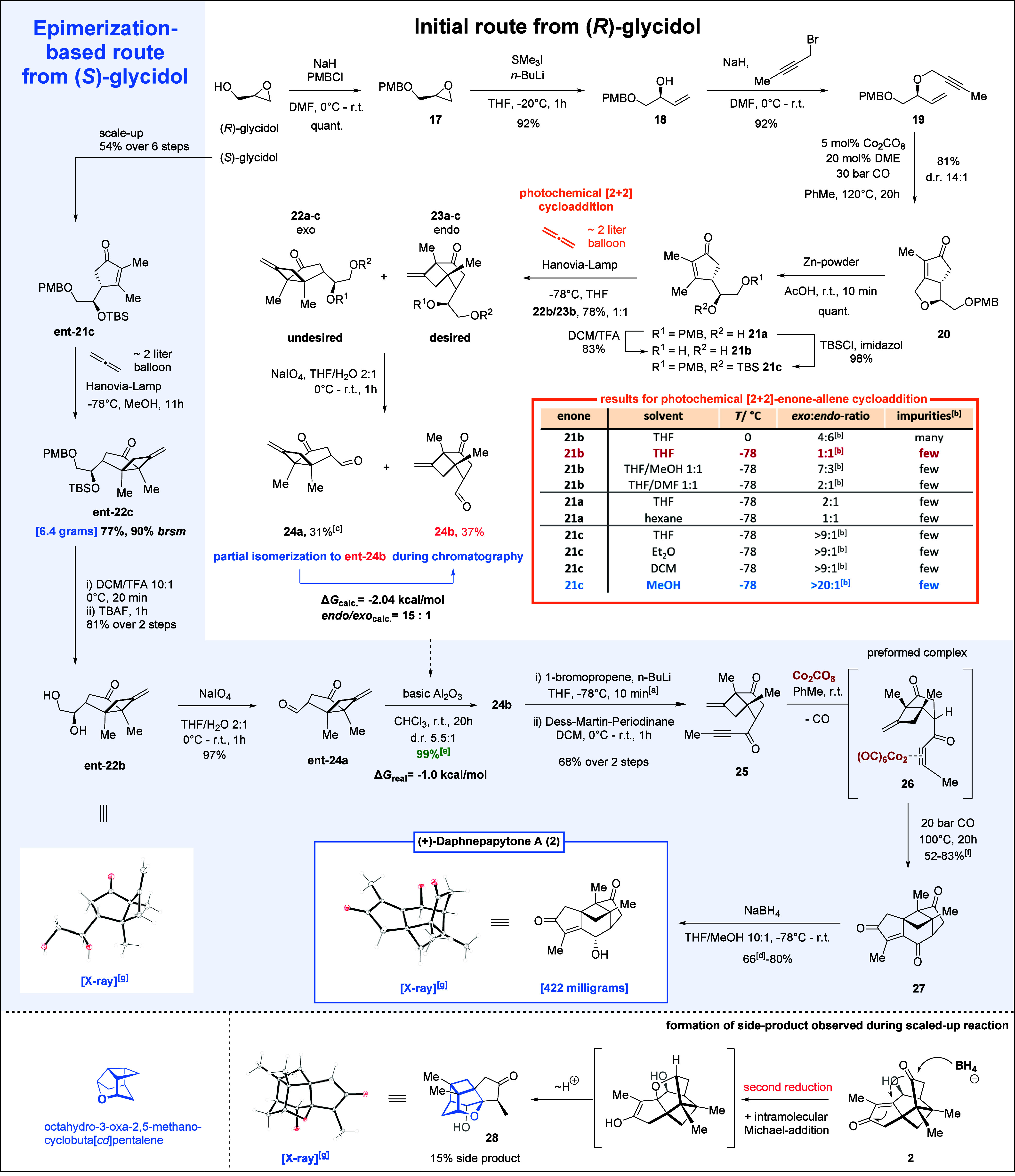 Figure 3
Figure 3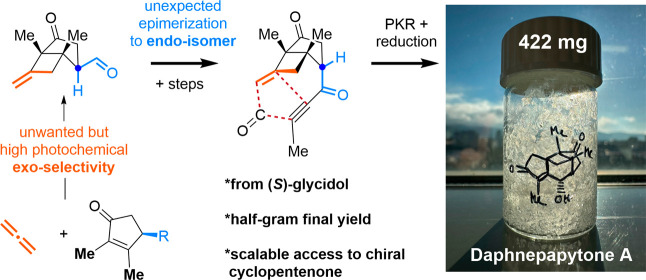 Figure 4
Figure 4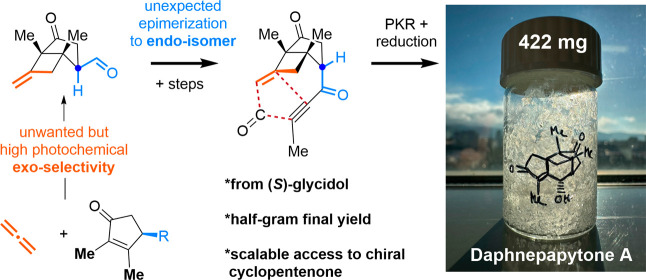 Figure 5
Figure 5- —Baden-W?rttemberg Stiftung10.13039/100008316
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Synthetic Organic Chemistry · Catalytic Alkyne Reactions · Synthesis and pharmacology of benzodiazepine derivatives
Daphnepapytone A is an unusual sesquiterpenoid that was first isolated from the dried stems of Daphne papyracea,? an evergreen shrub from East Asia found at altitudes between 700 and 3100 m. Extracts of this shrub have been shown to possess inhibitory activity against a protease of the Hepatitis C virus,? and daphnepapytone A specifically was found to have moderate α-glycosidase inhibitory activity (IC_50_ = 159 μM).? Only three years after its first isolation, its intriguing structure featuring a cyclopentenone-annelated tricyclo[4.3.0.0^3,9^]nonane cage has attracted the attention of organic chemists, leading to four published total syntheses (Scheme). ?−? ? ? All four routes are ex-chiral pool syntheses from (R)-carvone as the starting material, bringing with it the isopropenyl group with the correct stereoinformation. Cleavage of carvone via Eschenmoser–Tanabe fragmentation gave a precursor 1 that was converted into allene-yne 3 via an alcohol analogue thereof. Nay’s, Li/She’s, and Hanson’s syntheses follow a biomimetic approach, converting the allene-yne using a rhodium-catalyzed Pauson–Khand reaction (PKR) to access oleodaphnone? (5), a guaiane type sesquiterpene, as a photochemical precursor and proposed intermediate in the biosynthesis of daphnepapytone A.? Indeed, the intramolecular photochemical [2 + 2]-cycloaddition proceeds effectively, yielding the tetracyclic precursor 6 of daphnepapytone A which, following a CH-oxidation adjacent to the enone, is selectively reduced to the target compound. Stoltz followed a different approach to establish the central cyclobutane. In an unusual thermal allenone-ene-[2 + 2]-cycloaddition,? the cyclobutane is established, albeit with low selectivity toward the desired endoproduct 4. A subsequent Co_2_CO_8_-mediated PKR yields the tetracyclic cage 6 of daphnepapytone A, and the final compound is acquired in a similar oxidation–reduction sequence.?
In our synthesis plan (Schemei), we envisioned omitting the CH-oxidation of 6 featured in the previously published routes and directly using a propargylic alcohol 7 in a Pauson–Khand reaction to access the final compound. If the natural selectivity of propynyl-organometallic reagent cannot be tuned by other means, the propargylic alcohol could potentially be accessed via an asymmetric alkynation of an aldehyde. ?−? ? The aldehyde could be accessed from usually high yielding diol cleavage of 8. The cyclobutane featuring the methylidene group would be introduced in photochemical enone-allene-[2 + 2]-cycloaddition with head-to-head selectivity from precursor 9 accessed via the reductive cleavage of PKR-derived bicyclic ether 10. While asymmetric rhodium-catalyzed PKR is well-established and would not require to use a chiral enyne, reported catalyst loadings are typically >5 mol % ?,? and therefore it is expensive to access gram amounts of the compound. For that reason, we envisioned performing a diastereoselective cobalt-catalyzed PKR? of the easily accessible chiral enyne 11 that can be synthesized via attack of a sulfur-ylide? on protected (S)-glycidol and subsequent etherification. Most doubt for success was shown toward the desired endo-stereoselectivity of the intermolecular photochemical step (Schemeii), since no literature precedent exists for [2 + 2]-cycloadditions for the substitution pattern of 2,3,4- yet alone 3,4-substituted monocyclic cyclopentenones, e.g., 15. While bicyclic enones (12) have clear (but sometimes counterintuitive) stereochemical outcomes ?−? ? ? that can be empirically rationalized using a rule proposed by Wiesner,? data on monocyclic enones is sparse ?,? andfor the known examplesnot in favor of our synthetic plan. For example, an intermediate for the total synthesis of quadrone? was synthesized in over 80% yield via irradiation of a bicyclic enone, selectively furnishing the exo-tricycle 13. On the other hand, OTBS-substituted cyclopentenone yielded roughly 2:1 mixtures of exo:endo-products 14, without room for much tunability via solvent or substitution.? Unbothered by these odds and motivated by scientific curiosity, we deemed that applying Wiesner’s empirically rationalized model to our enone-system (15) would favor the endo-product due to higher interaction of the methyl-group at the assumed excited state’s (16) β-carbon with the residue R, an assumption that proved incorrect.
The synthesis (Scheme, white) began with protection of (R)-glycidol with PMBCl and subsequent sulfur-ylide attack to convert it to known chiral allylic alcohol 18, ?,? which was transformed into its corresponding propargylic ether 19. Initial attempts with TBDPS-protected glycidol suffered from diminished yields, presumably from silyl migration ?,? after epoxide ring opening. The chiral enyne 19 was subjected to a catalytic PKR with only 5 mol % cobalt octacarbonyl and 20 mol % dimethoxyethane as an additive? under 30 bar of CO pressure. Cyclopentenone 20 was generated in high yield and diastereoselectivity, and the minor isomer was separated via chromatography. To access the free methyl group of 21a, the tether was cleaved quantitatively after 10 min in a suspension of zinc in glacial acetic acid. ?,? While the sequence of a Pauson–Khand reaction with concomitant reductive cleavage of the ethereal tether is reported, ?,? our sequence using zinc circumvents the need for stoichiometric use of Co_2_CO_8._ Then, a thorough photochemical screening (see Supporting Information) with different protection (21a–c) at the vicinal diol followed, and no selectivity toward the desired endo-product could be achieved with solvent variation. At best, a ∼1:1 mixture of exo- and endo-products was formed at −78 °C in THF for deprotected diol substrate 21b. Higher temperatures converged toward 1:1 exo–endo mixtures with many inseparable side products. Increasing polarity while maintaining a low freezing point (e.g., THF/DMF 1:1) favored exo-product formation. The slightly greater steric bulk of substrate 21a further diminished the ratio of the wanted endo-bicycle (THF, 2:1). With even greater bulkiness introduced by TBS protection at the secondary alcohol (21c), the cycloaddition proceeded toward the unwanted exo-product 22c with high yield and selectivity. Capitulating on a mediocre endo-yield with substrate 21b, the synthesis proceeded with diol-mixture 22/23b, which was cleaved by using NaIO_4_. Luckily, the exo–endo mixture 24a/b of the resulting aldehyde was separable via a lengthy chromatography, during which an epimerization was noticed that partially converted the exo-epimer 24a into a mixture of the latter with the endo-enantiomer ent-24b so that full isomerization of this mixture would lead to a racemic compound rac-24b. This prompted us to investigate epimerization conditions, which were shown to be most effective in an NMR-tube with a small amount of basic alumina at the bottom, generating a 11:2 exo–endo mixture at equilibrium. The final relative stability of the isomeric aldehydes was qualitatively confirmed by DFT calculations, which assigned a ∼2 kcal difference in favor of the endo-isomer that was not quite met in reality. Since the [2 + 2]-cycloaddition toward the previously unwanted TBS-protected exo-isomer 22c proceeded selectively and with high yield, a new route (Scheme, blue shade) was devised starting from (S)-glycidol; this time, however, TBS-protected alcohol ent-21c was used in the photochemical step. This gave 77% (90% brsm) yield of >90% pure exo-photoadduct ent-22c, which was deprotected to yield the first solid compound ent-22b of this synthesis, confirming the structure via X-ray crystallography. After diol cleavage with periodate, epimerization of ent-24a at 1.5 g scale proceeded significantly more slowly than the NMR-scale screening, which was fixed by stirring the mixture, accelerating the isomerization from 5 days to overnight. Aldehyde 24b was reacted with propynyllithium to yield an inseparable diastereomeric 2:1 mixture of propargylic alcohols. Subjecting this mixture to Pauson–Khand conditions did not proceed cleanly, so the alcohols were oxidized using Dess-Martin periodinane to yield ynone 25 which, after preformation to cobalt-complex 26, underwent carbocyclization with yields ranging from 52% to 83% for different scales. For a similar step using substrate 4 (Scheme), Stoltz reports only a low yield due to a novel isomerization. Finally, reduction with NaBH_4_ at −78 °C in THF/MeOH furnished 422 mg of (+)-daphnepapytone A (2), yielding 2 orders of magnitude more substance than all previously reported procedures. ?−? ? ?
Interestingly, for the larger scale reaction, an over-reduced product 28 was isolated in 15% yield, which was previously not observed for a smaller scale. As visualized on structure 2, borohydride can attack the ketone from the convex side of the cage, bringing the resulting alcoholate in spatial proximity to the enone’s C3 and forcing the intramolecular Michael-addition. This results in the formation of yet another cage more complex than before, which can be described as a cyclopentenone-annelated octahydro-3-oxa-1H-2,5-methanocyclobuta-[cd]-pentalene, a motif not yet encountered in nature. Brief attempts to access 28 selectively via direct reduction of 2 have not yet been successful.
In summary, we achieved the de novo total synthesis of daphnepapytone A (2) through a sequence that features (1) efficient and convenient access to a chiral trisubstitued cyclopentenone via reductive cleavage of a Pauson–Khand-derived ether; (2) novel insights into the stereochemistry of photochemical [2 + 2]-cycloaddition to monocyclic enones, achieving high exo- selectivity through steric bulk; (3) serendipitous circumvention of exo-selectivity with mild and selective epimerization of the exo-aldehyde ent-24a to the endo-aldehyde 24b; and (4) a congested Pauson–Khand reaction of ynone 27 that generated a quaternary carbon at a cyclobutane with good yield. Since no data were available on the stereochemistry of [2 + 2] photocycloaddition to monocyclic cyclopentenones, we deem that illuminating this blind spot is of great use for future synthetic chemists. And last, being able to selectively access both endo- and exo-substituted bicyclo[3.2.0]heptanes (e.g., 24a/b) via one sequence could potentially open more efficient routes to other natural products. ?,?
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Huang S.-Z.Wang Q.Yuan J.-Z.Cai C.-H.Wang H.Mándi A.Kurtán T.Dai H.-F.Zhao Y.-X.Hexahydroazulene-2(1H)-one Sesquiterpenoids with Bridged Cyclobutane, Oxetane, and Tetrahydrofuran Rings from the Stems of Daphne papyracea with α-Glycosidase Inhibitory Activity J. Nat. Prod.202285131410.1021/acs.jnatprod.0c 0139434935371 · doi ↗ · pubmed ↗
- 2Wei Y.Yang J.Kishore Sakharkar M.Wang X.Liu Q.Du J.Zhang J.-J.Evaluating the inhibitory effect of eight compounds from Daphne papyracea against the NS 3/4A protease of hepatitis C virus Nat. Prod. Res.202034111607161010.1080/14786419.2018.151982530449158 · doi ↗ · pubmed ↗
- 3Gonzalez E. C.de la Torre Roehl I. M.Stoltz B. M.Concise total synthesis of the cage-like sesquiterpenoid (+)-daphnepapytone A Chem. Sci.20251625113811138510.1039/D 5SC 02952 J 40521111 PMC 12165289 · doi ↗ · pubmed ↗
- 4Martinez J. B.Hanson P. R.Total Synthesis of Daphnepapytone AJ. Org. Chem.20259042149771498310.1021/acs.joc.5c 0177441099155 PMC 12705262 · doi ↗ · pubmed ↗
- 5Pereira J.Casaretto N.Frison G.Nay B.Bio-inspired total synthesis of daphnepapytone A Chem. Sci.20251625113751138010.1039/D 5SC 02953 H 40417298 PMC 12096516 · doi ↗ · pubmed ↗
- 6Zhang Y.Yu P.Chen W.Li J.Liu K.Xie X.Li H.She X.Bioinspired Stereoselective Total Synthesis of the Caged Sesquiterpenoid Daphnepapytone A Org. Lett.202527215480548410.1021/acs.orglett.5c 0150940372148 · doi ↗ · pubmed ↗
- 7Li S.Wang X.Yang Y.Wu X.Zhang L.Discovering the Mechanisms of Oleodaphnone as a Potential HIV Latency-Reversing Agent by Transcriptome Profiling Int. J. Mol. Sci.2023248735710.3390/ijms 2408735737108519 PMC 10138910 · doi ↗ · pubmed ↗
- 8Hansen T. V.Skattebøl L.Stenstrøm Y.Synthetic efforts towards the protoilludenes. A formal synthesis of Δ7-protoilludene Tetrahedron 200359193461346610.1016/S 0040-4020(03)00480-0 · doi ↗