Dimerization of Allylbenzenes into Cyclolignans by a Metathesis-Oxidation Sequence
Kiryl Vasiutovich, Alexander A. Fadeev, Peter Čambal, Eliška Matoušová

TL;DR
A new chemical method combines metathesis and oxidation to efficiently produce cyclolignan compounds, which are important in natural product chemistry.
Contribution
A scalable one-pot metathesis-oxidation sequence for synthesizing cyclolignans with mechanistic insights.
Findings
A two-step sequence produces cyclolignans via Ru-catalyzed metathesis and oxidation.
Changing oxidation conditions yields benzyl styryl ketones instead of cyclolignans.
The oxidation mechanism involves single electron transfer and proton transfer steps.
Abstract
Allylbenzenes form dimeric cyclolignans in a two-step, one-pot sequence involving Ru-catalyzed olefin metathesis followed by iron- or acid-catalyzed oxidation with DDQ. Alternatively, performing the oxidation step under wet conditions leads to benzyl styryl ketones as the major products. The mechanistic investigation suggests that the oxidation process relies on the combination of single electron transfer, hydrogen atom transfer, and proton transfer steps. The developed metathesis-oxidation sequence enables a straightforward and scalable preparation of arylnaphthalene and aryltetralin types of cyclolignans, such as oleralignan B and its derivatives.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1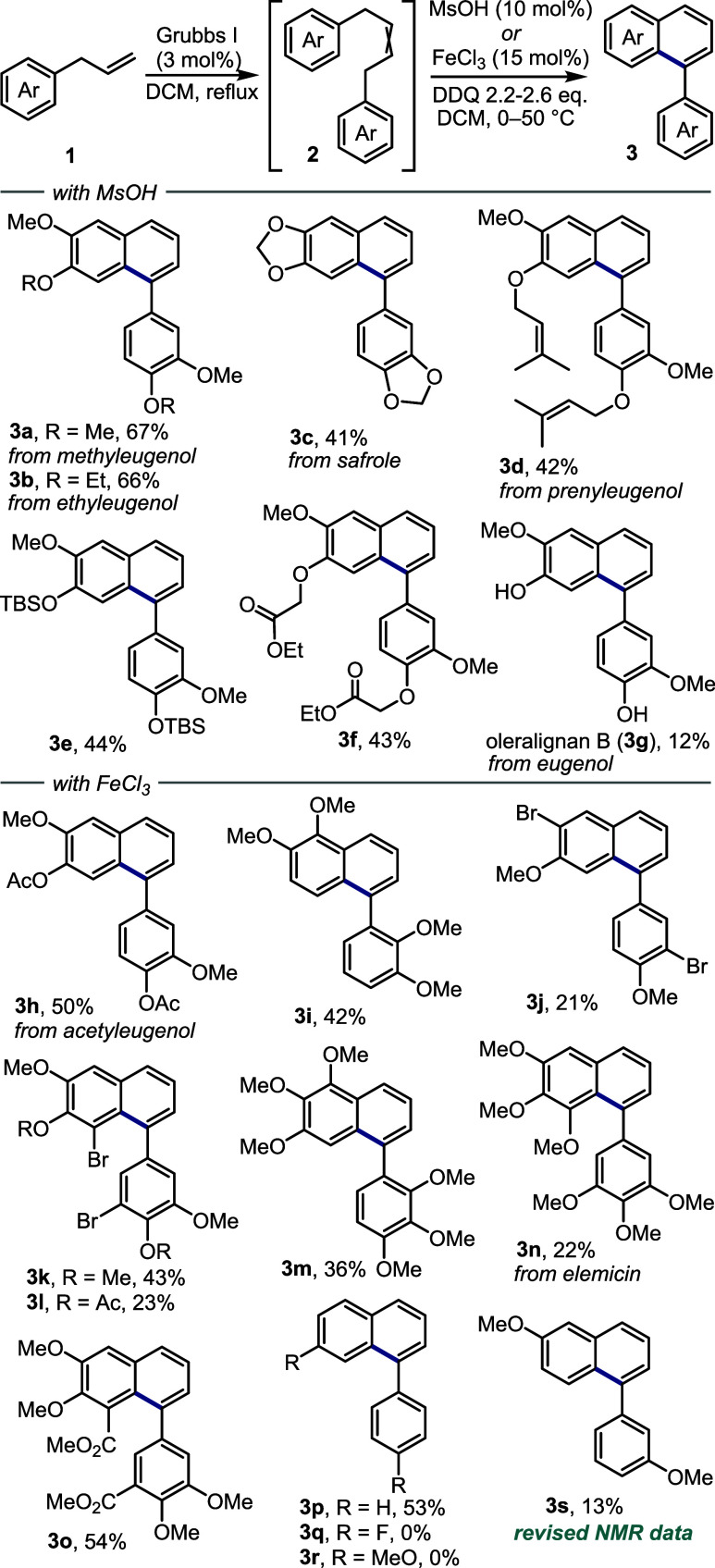 2
2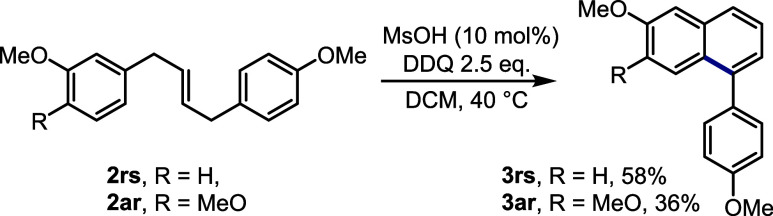 3
3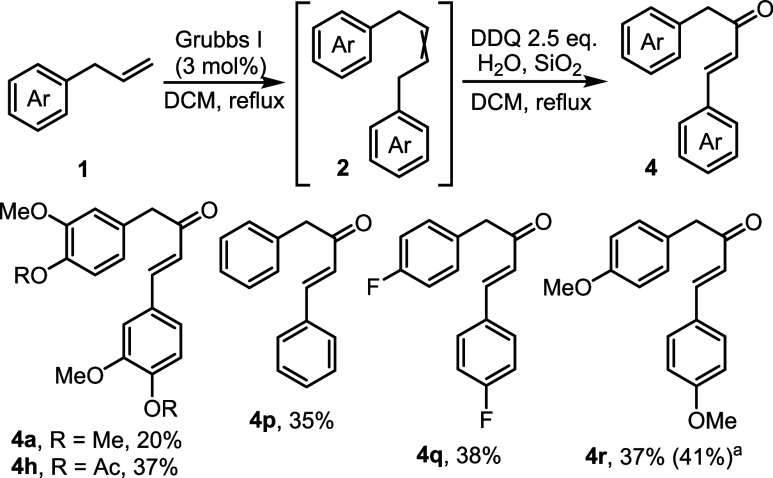 4
4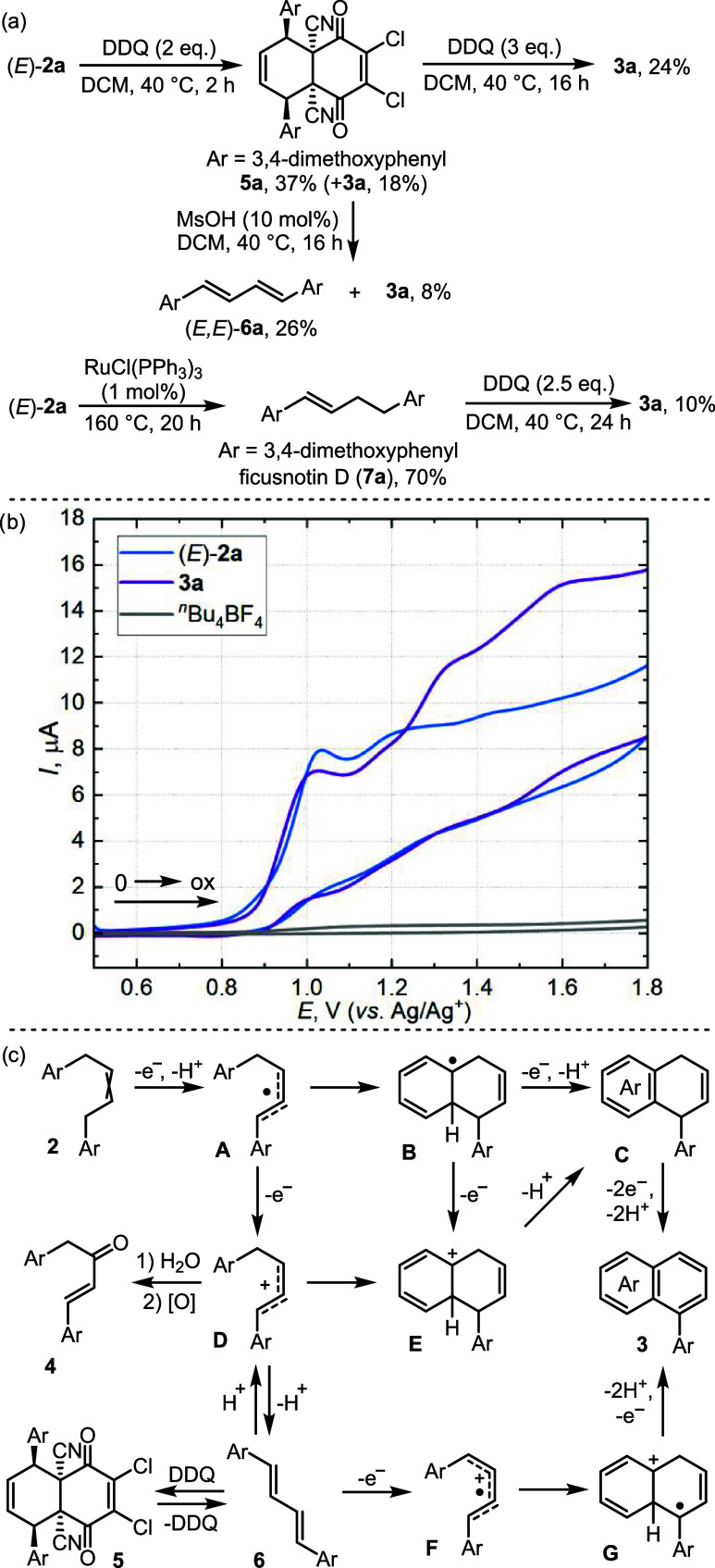 5
5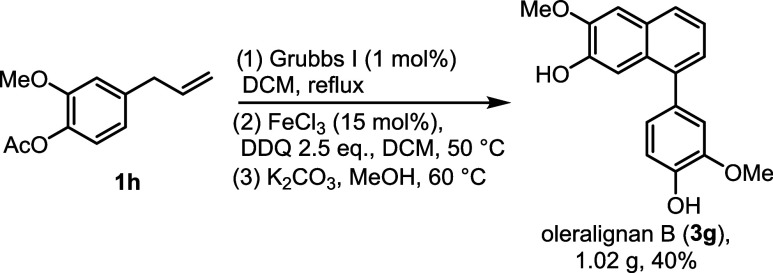 6
6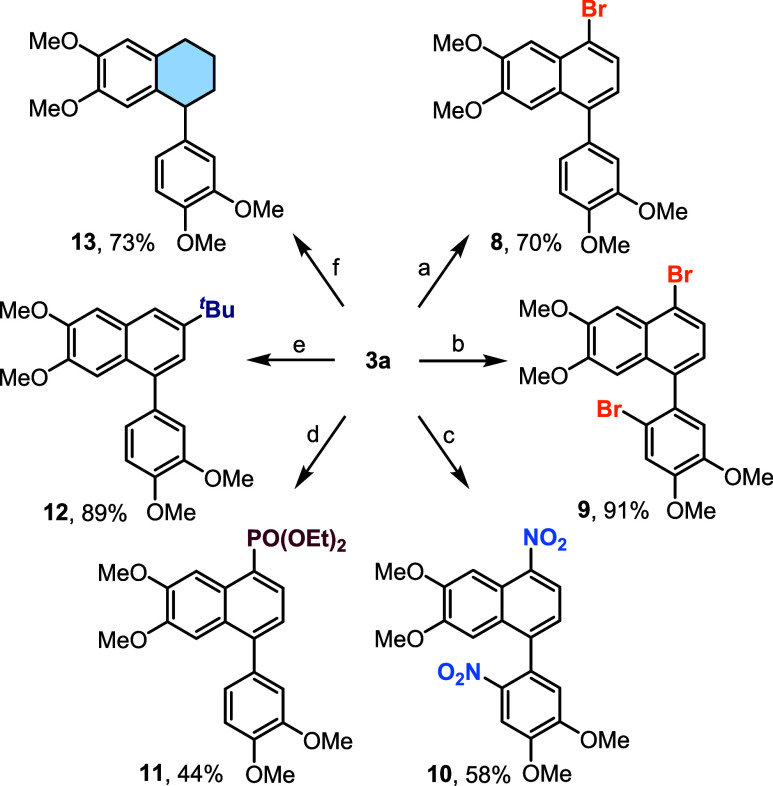 7
7| entry | oxidant (equiv) | additive (mol %) | yield of |
|---|---|---|---|
| 1 | DDQ (3) | 40 (34) | |
| 2 | chloranil (3) | 6 | |
| 3 | FeCl3 (3) | 15 | |
| 4 | FeCl3 (3), MnO2 (6) | 37 | |
| 5 | FeCl3 (0.15), DDQ (3) | 39 | |
| 6 | DDQ (3) | TFA (10) | 42 |
| 7 | DDQ (3) |
| 46 |
| 8 | DDQ (3) | MsOH (10) | 74 |
|
|
|
|
|
| 10 | DDQ (2) | MsOH (10) | 60 |
| 11 | DDQ (2.5) | MsOH (5) | 70 |
| 12 | DDQ (0.1), MnO2 (6) | MsOH (10) | 6 |
- —Univerzita Karlova v Praze10.13039/100007397
- —European Regional Development Fund10.13039/501100008530
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant-derived Lignans Synthesis and Bioactivity · Synthetic Organic Chemistry Methods · Magnolia and Illicium research
Introduction
Lignans represent a large family of natural bioactive small molecules of phenolic origin that are found in numerous plant and animal sources. ?−? ? ? ? Although the first known lignans have been recognized as a family since 1936,? new members are still being discovered. Lignans are considered to play a vital role in plant defense by expressing their antimicrobial, antifungal, antiviral, antioxidant, and insecticidal activities. ?−? ? ? ?,?−? ? At the same time, these properties have invigorated the research aiming at the preparation of lignans ?−? ? ? and their use as pharmaceuticals ?−? ? ? and food chemicals. ?,?
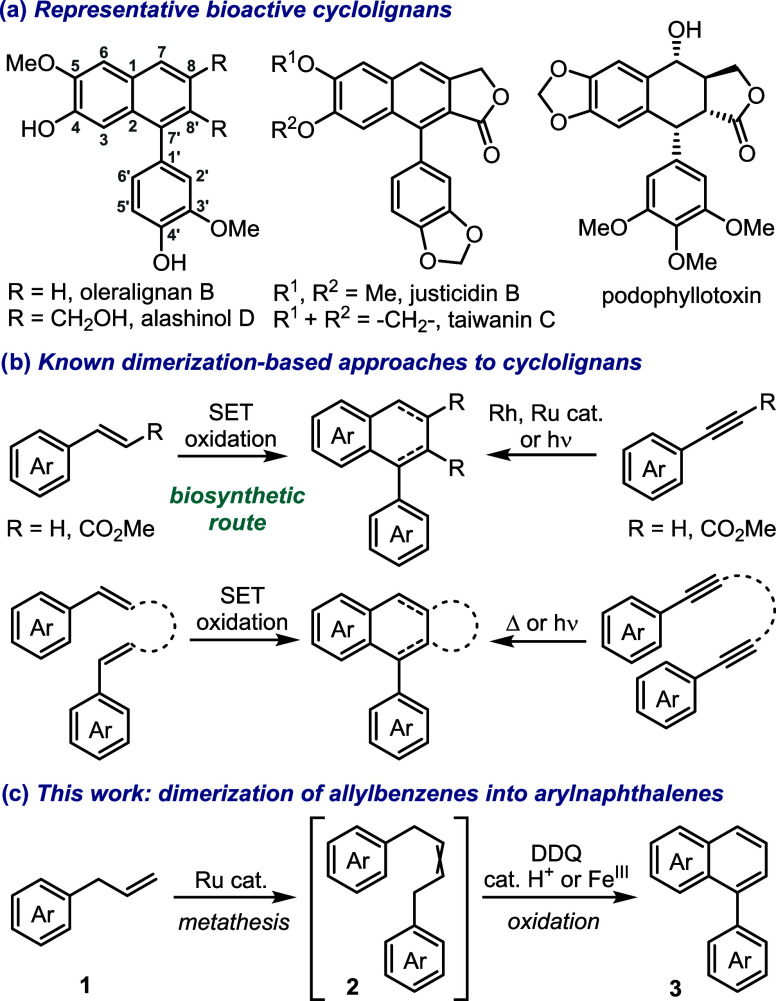
Cyclolignans, such as arylnaphthalenes and aryltetralins, are among the most prominent types of bioactive lignans (Schemea). ?,?,?,?,? For example, anti-inflammatory arylnaphthalenes oleralignan B and alashinol D have been recently isolated from Portulaca oleracea ? and Syringa pinnatifolia,? respectively. Arylnaphthalene lactones, such as justicidin B and taiwanin C, have been found in several plants over the decades and are known in particular for their activity against cancer cell lines. ?,? Podophyllotoxin and analogous aryltetralins from Podophyllum species exhibit a wide spectrum of biological activities and have been tailored into semisynthetic O-glycoside derivatives etoposide and teniposide for cancer treatment. ?,?
Cyclolignans and Their Synthesis from the Dimerization Precursors (Lignan Numbering is Shown)
Biosynthetically, the inherently dimeric structures of lignans originate from the dimerization of phenylpropanoid units.? This fact makes dimerization an effective strategy for the synthesis of lignans from a single building block, and cyclolignans are not an exception. Thus, chemical dimerization of styrenes resembles the biosynthetic dimerization of hydroxycinnamic acid derivatives ?,? and is typically initiated by one-electron oxidation to afford aryltetralins, ?−? ? aryltetralones, ?−? ? ? ? aryldihydronaphthalenes, ?,?−? ? and arylnaphthalenes, ?,?−? ? depending on the reaction conditions (Schemeb). The related dimerization of phenylacetylenes into arylnaphthalenes was first demonstrated by the thermal condensation of arylpropiolic acids ?−? ? and later extended in scope by the means of transition metal catalysis ?−? ? and the photodehydro-Diels–Alder reaction. ?−? ? However, controlling the chemo- and regioselectivity of the intermolecular dimerizations presents a challenge, and product mixtures are often formed. Tethering the reacting molecules in situ
?−? ? or as a separate reaction step ?−? ? ? ? ? ? ? ? markedly improves the effectiveness and the selectivity of the dimerization process. Nevertheless, the tethering approaches rely on specific functional groups in the reactants and create an additional ring that may not be desired in the final product. Besides that, the availability of the suitably substituted styrenes and phenylacetylenes is limited, and only a few can be obtained from renewable resources.
Conversely, the preparation of lignans by dimerization of allylbenzenes remains documented only by a handful of examples that do not include the synthesis of cyclolignans. ?−? ? Nonetheless, the increasing availability of allylbenzenes from renewable materials, including lignin and essential oils, makes these arenes attractive building blocks for organic synthesis. We suggested that dimerization of allylbenzenes 1 into 1,4-diaryl-2-butenes 2 by olefin metathesis could serve as a traceless tether approach enabling the subsequent oxidative annulation of 2 into arylnaphthalene lignans 3, such as oleralignan B and its congeners (Schemec). At the same time, we expected that the strain induced by the double bond in 2 would prevent the 8-membered ring formation observed earlier in the oxidation of electron-rich 1,4-diarylbutanes. ?−? ? Notably, alkenes 2 are common byproducts in the synthesis of other valuable compounds via olefin cross-metathesis, ?−? ? and harnessing the synthetic potential of these alkenes could reduce the waste generated with implementing such protocols. Lastly, the synthetic sequence described herein is scalable and shorter than most of the existing routes that do not rely on dimerization to access analogous cyclolignans. ?−? ? ?,?,?−? ? ? ? ? ? ? ? ?
Results and Discussion
Knowing that one-electron oxidants, such as DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) and iron(III) chloride, are particularly effective in annulation reactions, ?,?,? we tested these reagents in the oxidation of E-alkene 2a, which was in turn obtained by the self-metathesis of methyleugenol using Grubbs I catalyst. Gratifyingly, treating E-(2a) with 3 equiv of DDQ in dichloromethane afforded the desired annulation product 3a in 40% yield at 40 °C and in 34% yield at 25 °C (Table). Chloranil was considerably less effective than DDQ and gave only 6% of 3a. Likewise, a rapid formation of 3a was observed at 25 °C with FeCl_3_ as the sole oxidant (15% yield), with FeCl_3_ and MnO_2_ as the co-oxidant (37% yield), and using the FeCl_3_-DDQ system (39% yield). Next, we screened the performance of organic acids as additives. The oxidation of 2a with DDQ in the presence of 10 mol % of trifluoroacetic or p-toluenesulfonic acid improved the yield of 3a only to 42% and 46%, respectively. In contrast, the addition of methanesulfonic acid led to a nearly 2-fold increase in the product yield (74%), even when the amount of DDQ was reduced to 2.5 equiv. However, lowering the amount of DDQ to 2 equiv or the amount of acid to 5 mol % decreased the yield to 60% and 70%, respectively. The attempted use of DDQ in a catalytic amount with MnO_2_ as the terminal oxidant gave only a trace amount of 3a.
1: Screening of the Aromatization Conditions
With the optimized conditions of the annulation reaction in hand, we paired the metathesis with the annulation in the one-pot process and explored the applicability of this sequence. Thus, subjecting methyleugenol to metathesis using a Grubbs I catalyst and treating the reaction mixture with DDQ and MsOH afforded lignan 3a in 67% overall yield (Scheme). Likewise, natural eugenol analogs such as ethyleugenol, safrole, and prenyleugenol provided lignans 3b–3d in 66%, 41%, and 42% yields, respectively. Products 3e and 3f were obtained in similar yields from silylated eugenol 1e (44%) and chloroacetate derivative 1f (43%). Presumably, the yields of 3c–3f were affected by the cleavage of the aryl ethers during the annulation step, leading to decomposition. Nonetheless, unsubstituted eugenol permitted the first synthesis of oleralignan B (3g), although in only 12% yield (the improved synthesis is described in Scheme).
Scope of the Lignans Prepared by the One-Pot Metathesis-Oxidation Sequence (Overall Isolated Yields are Given)
However, when we tried to apply the protocol to alkenes of different substitution character, such as acetyleugenol, allylbenzene, and 2,3-dimethoxyallylbenzene, the desired products were not detected after the annulation step. Complex product mixtures were formed, in which adducts 5 (vide infra) were observed by ^1^H and ^13^C NMR. In these cases, extending the reaction time and introducing more methanesulfonic acid led to only the decomposition of 5. Nevertheless, using the FeCl_3_-DDQ system for annulation gave better results. Thus, acetyleugenol afforded a diacetate derivative of oleralignan B (3h) in 50% yield. Next, 2,3-dimethoxyallylbenzene yielded 42% of contiguously substituted arylnaphthalene 3i. The cyclization of intermediate 2j arising from 3-bromo-4-methoxyallylbenzene gave regioselectively the product of para attack with respect to the position of bromine, albeit in low yield (3j, 21%). At the same time, brominated eugenol derivatives 1k and 1l provided the cyclization products 3k (43% yield) and 3l (23% yield) resulting from the ortho attack with respect to the bromine atom, evidently due to the activation of this position by the para-methoxy group. Electronically rich 2,3,4-trimethoxyallylbenzene and 3,4,5-trimethoxyallylbenzene (elemicin) furnished lignans 3m and 3n in 36% and 22% yields, respectively. Replacing the methoxy group in position 3 of elemicin with a methoxycarbonyl group more than doubled the yield of the product 3o (54%). Electronically neutral allylbenzene gave 1-phenylnaphthalene (3p) in 53% yield. Surprisingly, para-substituted allylbenzenes 1q and 1r did not give the desired products neither in the presence of MsOH nor FeCl_3_. Presumably, the positive mesomeric effect of the individual para-methoxy and para-fluoro substituents induces an electronic mismatch that hampers the cyclization step. Lastly, meta-methoxyallylbenzene afforded lignan 3s (13% yield), the NMR data of which were revised to resolve the inconsistencies in the previous reports describing its preparation. ?,?,?
To uncover whether unsymmetrical alkenes produced by cross-metathesis could selectively undergo the annulation reaction, we prepared alkenes 2rs and 2ar and subjected them to the standard acid-catalyzed annulation protocol. As shown in Scheme, in both cases, the cyclization occurred regioselectively at the free para position to the methoxy group, giving rise to arenes 3rs (58%) and 3ar (36%).
Annulation of the Unsymmetrical Alkenes Obtained by the Cross-Metathesis
The inability of metathesis products 2q and 2r to cyclize motivated us to investigate the intermolecular reactivity of the generated intermediates. Aiming to trap the presumably cationic species with water, we performed the annulation step with DDQ in the presence of wet silica gel without any other additives. ?,? As shown in Scheme, when 2a was generated by the metathesis of 1a and subjected to oxidation under these conditions, benzyl styryl ketone 4a was isolated in 20% yield together with lignan 3a (33% yield). As expected, higher yields of enones 4 were observed starting from alkenes bearing less electron-rich aromatic rings, and only trace amounts of lignans 3 were detected. Thus, enones 4h and 4p–4r were obtained in 35–38% yields exclusively as the trans-isomers (no other isolable products were attained). The yield of 4r was increased in the presence of iron trichloride as an additive, albeit only to 41%.
One-Pot Synthesis of Benzyl Styryl Ketones from Allylbenzenes
The mechanism of the annulation process was then probed by studying the possible reaction intermediates and cyclic voltammetry (CV) profiles. Thus, when the reaction of (E)-2a with 2 equiv of DDQ was stopped after 2 h, 3a was obtained in 18% yield together with Diels–Alder adduct 5a as the major reaction product isolated in 37% yield (Schemea). The subsequent treatment of 5a with DDQ under the same conditions provided 3a in a 24% yield. Similarly, methanesulfonic acid catalyzed the cleavage of 5a leading to trans-trans-diene 6a (26%) and 3a (8%). Although both 5a and 6a were also detected during the oxidation of (E)-2a with DDQ in the presence of MsOH, no isolable intermediates were found when FeCl_3_ was used as the additive. Treating (E)-2a in DCM with 10 mol % MsOH at 40 °C had no effect. To investigate the possibility of the double bond migration prior the cyclization, isomeric alkene 7a (ficusnotin D) was prepared by Ru-catalyzed isomerization of (E)-2a ? and subjected to oxidation with DDQ. Diene 6a was not detected during the reaction course, and 3a was the only observed product, albeit in only 10% yield. Finally, the CV profiles of (E)-2a and 3a showed that both compounds are irreversibly oxidized at potentials slightly above 1.0 V vs. Ag/Ag^+^ (Schemeb; see Supporting Information for details). The small difference in the oxidation potentials of these molecules indicates that further oxidation reactions may occur after the annulation step, negatively affecting the product yield. The one-electron reduction potential of Fe^III^/Fe^II^ (−0.25 V vs. Ag/Ag^+^) is considerably lower than that of DDQ/DDQ^●–^ (0.22 V vs. Ag/Ag^+^), suggesting that DDQ likely initiates the oxidation also when FeCl_3_ is present in the mixture.
Control Experiments (a), CV Profiles of Compounds (E)-2a and 3a (b), and the Proposed Reaction Mechanism (c)
Based on the above experimental observations and the literature data, we propose the mechanistic pathways of the oxidative annulation, as shown in Schemec. Initially, abstraction of a hydrogen atom from the benzylic position of alkene 2 by DDQ leads to the delocalized radical A. In the case of alkenes 2 equipped with electron-rich aromatic rings (e.g. 2a, 2m, 2n), single electron oxidation followed by deprotonation may be the preferred pathway to this intermediate. After that, if a radical-accepting aromatic ring is present in A, the direct radical cyclization could form intermediate B, and the ensuing hydrogen abstraction steps would consecutively give 1,4-dihydronaphthalene C and the final aromatic product 3.? Alternatively, single electron oxidation of radical A to cation D paves the way for the ionic reactivity. ?,? First, cation D could undergo Friedel–Crafts-type cyclization leading to product 3 through intermediates E and C. Second, elimination of a proton from cation D leads to diene 6 that can react with DDQ to form Diels–Alder adduct 5 or undergo oxidation into 3, possibly through delocalized radical-cation intermediates F and G. Third, hydration of D followed by oxidation of the resulting alcohol leads to thermodynamically favorable trans-enone 4. ?,?
Overall, the mechanism of the oxidative annulation of 2 with DDQ depends on the substitution pattern of the aromatic rings and the additive used. The acid additive does not affect alkene 2, but it catalyzes the retro-Diels–Alder cleavage of adduct 5, reversibly protonates diene 6, and may also promote shifts and isomerizations of the double bonds in the reaction intermediates. In contrast, FeCl_3_ alone can convert 2 into 3, yet using it as the stoichiometric oxidant dramatically decreases the yield of 3, likely due to unwanted intermolecular Scholl reactions.? Presumably, the catalytic amount of FeCl_3_ facilitates the cyclization step. Besides that, FeCl_3_ can act as a source of hydrochloric acid.
To demonstrate the applicability of the developed protocol, we improved and upscaled the synthesis of bioactive natural product oleralignan B (Scheme). Starting from inexpensive acetyleugenol (1h), we avoided exposing the labile phenolic rings to the oxidative conditions of the annulation process. Thus, self-metathesis of 1h on a gram scale showed full conversion even with the loading of the Grubbs I catalyst reduced to 1 mol %. Engaging the formed alkene 2h directly into the oxidation step using the FeCl_3_-DDQ system gave diacetate 3h in 43% yield, and 84% of the reduced DDQ was recovered (the yield of 3h on this scale was likely lowered by the additional extraction step during workup). Finally, base-mediated hydrolysis of 3h provided oleralignan B in 95% yield (1.02 g, 40% overall from 1h).
Gram-Scale Preparation of Oleralignan B from Acetyleugenol
Additionally, using 3a as a stable model substrate, we explored the options for a selective postfunctionalization of the oleralignan B core with substituents that are difficult to introduce through the annulation process. First, selective bromination was achieved by controlling the reaction temperature and the amount of N-bromosuccinimide (NBS) used as the bromine source, allowing to prepare either bromoarene 8 in 70% yield or dibromoarene 9 in 91% yield (Scheme). However, nitration was more difficult to control; therefore, only 7,6′-dinitroderivative 10 was obtained in 58% yield. Phosphorylation with triethyl phosphite under photoredox conditions? exhibited monoselectivity toward position 7 leading to phosphonate 11, albeit only in 44% yield due to an incomplete conversion of 3a. In contrast to the reactions with heteroatom-based electrophiles, Friedel–Crafts monoalkylation with tert-butanol in the acidic medium resulted in the substitution of position 8 and furnished alkylarene 12 in 89% yield. Finally, ring-selective Birch reduction? afforded aryltetralin 13 in 73% yield. Overall, the reactivity of lignan 3a follows the general aromatic substitution patterns, with a high preference for the attack on the central aromatic ring.
Synthetic Modification of Lignan 3a
Conclusions
In summary, we found that both symmetrical and unsymmetrical 1,4-diaryl-2-butenes undergo oxidation into arylnaphthalenes with DDQ in the presence of catalytic amounts of methanesulfonic acid or iron(III) chloride. At the same time, oxidation of such alkenes with DDQ in the presence of water and silica gel leads preferentially to benzyl styryl ketones. Although modest yields are typical for these transformations, the reactions can be performed in a one-pot manner directly after preparation of the starting alkenes by the olefin metathesis of abundant allylbenzenes. By relying on this reaction sequence, the first synthesis of oleralignan B was accomplished on a gram scale. Additionally, we demonstrated that the oleralignan B core allows for a selective synthetic modification, including bromination, nitration, phosphorylation, alkylation, and reduction.
Safety Statement
Caution! DDQ can be decomposed in the presence of water and potentially release highly toxic hydrogen cyanide. Reactions involving large amounts of DDQ under wet conditions and the subsequent aqueous workup should be conducted in a fume hood. Bromine is a volatile, corrosive, and toxic liquid. Chemical-resistant gloves and safety goggles are essential for a safe handling. Toxic and corrosive hydrogen bromide vapors are released during bromination. A fume hood with proper ventilation is necessary to avoid a direct inhalation or skin contact. Sodium vigorously reacts with water. Exposure of sodium to moist air should be avoided. Sodium metal reacts violently with ethanol and releases flammable hydrogen gas. Therefore, the reaction should be conducted in a fume hood, especially on a large scale.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Calvo-Flores, F. G. ; Dobado, J. A. ; Isac-García, J. ; Martín-Martínez, F. J. Lignin and Lignans as Renewable Raw Materials: Chemistry, Technology and Applications; John Wiley & Sons, 2015.
- 2Pan J.-Y.Chen S.-L.Yang M.-H.Wu J.Sinkkonen J.Zou K.An Update on Lignans: Natural Products and Synthesis Nat. Prod. Rep.200926101251129210.1039/b 910940 d 19779640 · doi ↗ · pubmed ↗
- 3Saleem M.Kim H. J.Ali M. S.Lee Y. S.An Update on Bioactive Plant Lignans Nat. Prod. Rep.200522669671610.1039/b 514045 p 16311631 · doi ↗ · pubmed ↗
- 4Teponno R. B.Kusari S.Spiteller M.Recent Advances in Research on Lignans and Neolignans Nat. Prod. Rep.20163391044109210.1039/C 6NP 00021 E 27157413 · doi ↗ · pubmed ↗
- 5Landete J. M.Plant and Mammalian Lignans: A Review of Source, Intake, Metabolism, Intestinal Bacteria and Health Food Res. Int.201246141042410.1016/j.foodres.2011.12.023 · doi ↗
- 6Turner E. E.Hirst E. L.Peat S.Haworth R. D.Baker W.Linstead R. P.Cook J. W.Organic Chemistry Annu. Rep. Prog. Chem.193633022838210.1039/ar 9363300228 · doi ↗
- 7Zálešák F.Bon D. J.-Y. D.Pospíšil J.Lignans and Neolignans: Plant Secondary Metabolites as a Reservoir of Biologically Active Substances Pharmacol. Res.201914610428410.1016/j.phrs.2019.10428431136813 · doi ↗ · pubmed ↗
- 8Cui Q.Du R.Liu M.Rong L.Lignans and Their Derivatives from Plants as Antivirals Molecules 202025118310.3390/molecules 2501018331906391 PMC 6982783 · doi ↗ · pubmed ↗