Role of Ferredoxin 1 (FDX1) in cancer and its therapeutic potential
Fen He, Hongyan Zhao, Ruixin Gao, Maoyou Lu, Siqi Wang, Yu Chen, Lijun Peng, Jiliang Xia

TL;DR
FDX1, a mitochondrial protein, is linked to cancer progression and immune responses, and may serve as a potential biomarker and therapeutic target.
Contribution
This review highlights FDX1's dual roles in cancer and its potential as a mediator of cuproptosis and immunotherapy target.
Findings
FDX1 expression is dysregulated in various cancers, with both upregulation and downregulation observed.
FDX1 mediates cuproptosis, a copper-dependent cell death mechanism, suggesting tumor-suppressive roles.
FDX1 correlates with tumor immunity and may act as a biomarker for cancer prognosis and immunotherapy response.
Abstract
Ferredoxin 1 (FDX1) is a small iron-sulfur (Fe–S) cluster protein localized to the mitochondria. It functions as an electron carrier in diverse metabolic pathways and is critically involved in regulating protein lipoylation Accumulating evidence indicates that FDX1 expression is frequently dysregulated across various cancer types. Its expression is significantly associated with cancer progression, prognosis, and tumor immune responses, suggesting its potential as a biomarker for cancer diagnosis, prognostic evaluation, and immunotherapy response prediction. Transcription factors, epigenetic modifications, and non-coding RNAs (ncRNAs) contribute to the aberrant expression of FDX1 in cancer cells. Mechanistic studies reveal that FDX1 protein influences cancer progression by modulating oncogenic signaling pathways, metabolic reprogramming, and tumor immunity; Notably, FDX1 serves as a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
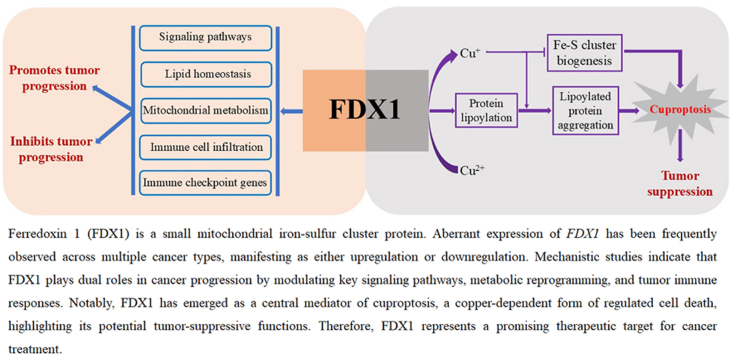 Figure 1
Figure 1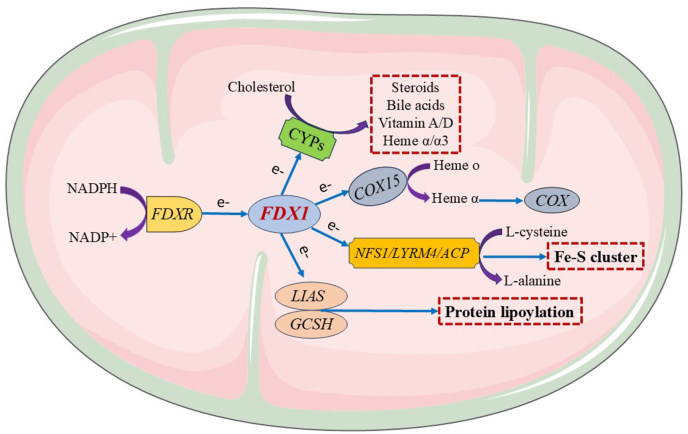 Figure 2
Figure 2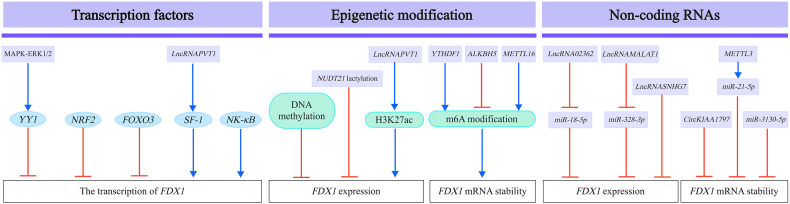 Figure 3
Figure 3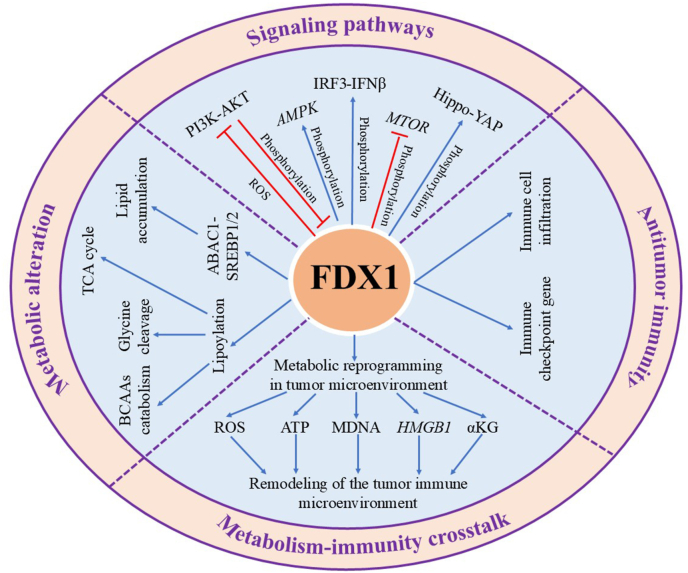 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetalloenzymes and iron-sulfur proteins · Ferroptosis and cancer prognosis · Cancer, Hypoxia, and Metabolism
Introduction
Ferredoxins (FDXs) are evolutionarily conserved iron-sulfur (Fe–S) cluster-containing proteins localized in the mitochondria, where they serve as electron transfer mediators in various metabolic pathways. In mammals, the two ferredoxin isoforms, FDX1 and FDX2, share 43% sequence identity and possess highly similar structural characteristics.1, 2, 3 Evidence increasingly supports a unique role for FDX2 in Fe–S cluster biogenesis.1 By contrast, FDX1 participates in a diverse range of biological functions, including protein lipoylation, Fe–S cluster biosynthesis, steroid and bile acid synthesis, heme α/α3 formation, vitamin A and D metabolism, and copper ion reduction.1
FDX1 expression is dysregulated in various cancers, such as gastric cancer,4 hepatocellular carcinoma (HCC),5 breast cancer,6 colorectal cancer,7 clear cell renal cell carcinoma (ccRCC),8 and glioma.9 This aberrant expression is significantly associated with cancer progression, patient prognosis, and tumor immune responses, suggesting that the FDX1 gene may serve as a promising biomarker for cancer diagnosis, prognostic evaluation, and immunotherapy response prediction. Multiple factors contribute to the altered FDX1 expression in these malignancies, such as regulation by transcription factors,7^,^10, 11, 12, 13 epigenetic modifications,7^,^14, 15, 16, 17, 18 and non-coding RNAs (ncRNAs).19, 20, 21, 22, 23, 24 The FDX1 protein plays a critical role in key cancer-related processes, including cell proliferation, drug resistance, radioresistance, invasion, and metastasis across multiple tumor types.8^,^15^,^25, 26, 27 Mechanistically, FDX1 modulates cancer progression by regulating oncogenic signaling pathways,5^,^28, 29, 30 metabolic reprogramming,31, 32, 33 and tumor immunity.34^,^35 Under conditions of copper overload, FDX1 reduces Cu^2+^ to the toxic Cu ^+^ form, promoting the aggregation of lipoylated proteins and inhibiting Fe–S cluster biogenesis, which subsequently induces cuproptosis—a copper-dependent form of programmed cell death.36 Therefore, FDX1 exerts a dual role in cancer, acting either as a promoter or suppressor of cancer progression depending on the cancer type. Given its central involvement in oncogenesis and cuproptosis, several anticancer strategies targeting FDX1 directly or indirectly have been developed, showing substantial therapeutic potential in various malignancies.11^,^37, 38, 39, 40 This review aims to summarize the current understanding of FDX1 gene dysregulation and its regulatory mechanisms in cancer, outlines the molecular pathways through which FDX1 protein influences cancer progression, and discusses the therapeutic potential of targeting FDX1. Furthermore, it highlights future research directions for FDX1 gene studies and the development of novel anticancer therapeutic strategies.
Physiological functions of FDX1 in normal cells
The human FDX1 gene is located on chromosome 11q22.3 and consists of two exons encoding a 184-amino-acid protein with a molecular weight of 19.4 kDa. The FDX1 protein participates in multiple biological processes in mammalian cells [Figure 1]. Its canonical function lies in steroid biosynthesis, where FDX1 receives electrons from NADPH via ferredoxin reductase (FDXR), an NADPH-dependent flavoprotein, and transfers these electrons to mitochondrial cytochrome P450 enzymes (CYPs), thereby initiating steroidogenesis.41^,^42 The mitochondrial FDX1-CYP system not only drives steroid biosynthesis but also regulates the production of bile acids, heme α/α3, and vitamins A/D^42^. FDX1 depletion reduces both the expression and activity of cyclooxygenase (COX), a heme α/α3-dependent enzyme, in human cells, underscoring its regulatory role in COX function.42^,^43 Mechanistic studies indicate that FDX1 regulates COX by delivering electrons to cytochrome c oxidase assembly homolog 15 (COX15), the heme α synthase.42 FDX1 also interacts with the cysteine desulfurase complex involved in Fe–S cluster biogenesis, which consists of cysteine desulfurase, LYR motif-containing 4, and acyl carrier protein.2 Within this complex, FDX1 donates electrons to facilitate the conversion of l-cysteine to l-alanine, leading to Fe–S cluster generation.2 As an upstream regulator of cellular protein lipoylation, a mitochondrial lipid-dependent post-translational modification, FDX1 interacts with lipoyl synthase (LIAS) and facilitates its association with the lipoyl carrier protein glycine cleavage system H (GCSH).33 Lipoylation is critical for the enzymatic activity of four mitochondrial multienzyme complexes: pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, branched-chain α-ketoacid dehydrogenase, and the glycine decarboxylase complex.33 Thus, FDX1 plays a central role in the tricarboxylic acid (TCA) cycle, branched-chain amino acid (BCAA) catabolism, and the glycine cleavage pathway.33 FDX1-mediated lipoylation not only supports mitochondrial metabolism but also generates substrates required for cuproptosis. Under conditions of copper overload, FDX1 reduces Cu^2+^ to Cu^+^, which promotes the aggregation of lipoylated proteins and inhibits Fe–S cluster biogenesis, ultimately inducing cuproptosis.36 Consequently, FDX1 acts as a key mediator of cuproptosis. Overall, FDX1 functions as an electron transfer protein and reductase that participates in diverse metabolic pathways and plays a role in cuproptosis.Figure 1The physiological functions of FDX1. FDX1 receives electrons from NADPH via FDXR and subsequently participates in various cellular processes by donating these electrons to downstream mitochondrial acceptors. It donates electrons to mitochondrial cytochrome P450 enzymes (CYPs), thereby initiating the biosynthesis of steroids, bile acids, vitamin A and D, and heme a/a3. Furthermore, FDX1 modulates COX activity by delivering electrons to COX15, the heme α synthase essential for COX assembly and function. In addition, FDX1 interacts with the NFS1-LYRM4-ACP complex to facilitate the conversion of l-cysteine to l-alanine and support Fe–S cluster biogenesis via electron transfer. FDX1 also functions as an upstream regulator of protein lipoylation by binding to LIAS and enhancing its functional interaction with the lipoyl carrier protein GCSH. ACP: Acyl carrier protein; COX: Cyclooxygenase; COX15: Cytochrome c - oxidase assembly homolog 15; FDX1: Ferredoxin 1; FDXR: Ferredoxin reductase; GCSH: Glycine cleavage system H-protein; LIAS: Lipoyl synthase; LYRM4: LYR motif containing 4;.NADPH: Nicotinamide adenine dinucleotide phosphate; NFS1: Nitrogen fixation 1.Figure 1
Aberrant expression of FDX1 and its regulatory mechanisms in human cancer
Given the critical role of the FDX1 gene in multiple cellular processes, normal cells maintain tight regulation of its expression. However, in numerous cancer types, FDX1 expression is frequently dysregulated at both the mRNA and protein levels. This dysregulation is significantly associated with either poor or favorable prognosis, underscoring the potential of FDX1 as a prognostic biomarker [Table 1]. Several regulatory mechanisms contribute to aberrant FDX1 expression across different cancers, including transcription factors, epigenetic modifications, and ncRNAs [Figure 2].Table 1. Overview of FDX1 expression in cancer tissues and cell lines.Table 1FDX1 expressionCancer typeSample typeDetection methodBiological functionReferencesUpregulationOvarian cancerTumor tissues, cell linesTCGA, qRT-PCR, and IHCPromotes progression and drug resistance25,28Multiple myelomaTumor tissues, cell linesGEO and qRT-PCRExhibits strong diagnostic value and promotes disease progression by modulating immune-related pathways110Stomach adenocarcinomaTumor tissuesGEO, TCGA, and IHCActs as a diagnostic marker and is positively associated with favorable outcomes16,111GliomaTumor tissuesTCGA, WB, and IHCPositively associated with poor prognosis, immune cell infiltration, and immune checkpoint genes and mediates cuproptosis34,45,112Uterine corpus endometrial carcinomaTumor tissuesTCGAPositively associated with Poor prognosis and immune-related genes15OsteosarcomaTumor tissues and cell linesGEO and qRT-PCRPositively associated with poor outcome and immune cell infiltration and promotes migration113DownregulationHepatocellular carcinomaTumor tissuesTCGA, GEO, IHC, qRT-PCR, and WBPositively associated with favorable outcomes, inhibits tumor growth and metastasis, and mediates cuproptosis5,44Papillary thyroid carcinomaTumor tissuesTCGA, GEO, and IHCNegatively associated with tumor recurrence and positively associated with immune cell infiltration114Colorectal cancerTumor tissuesTCGA and IHCPositively ssociated with favorable clinical outcomes and immune cell infiltration, and mediates cuproptosis.7,96Clear cell renal cell carcinomaTumor tissues, cell linesGEO, TCGA, IHC, and WBPositively associated with favorable outcomes and immune infiltration, inhibits tumor growth and metastasis, and mediates cuproptosis8,45,115Non-small cell lung cancerTumor tissues, plasmaTCGA, GEO, IHC, and ELISAActs as a diagnostic marker and is positively associated with favorable outcomes, inhibits cell proliferation, migration, and invasion97,116,117Breast cancerTumor tissuesTCGAPositively associated with favorable prognosis, induces malignant progression and immune suppression45,118Diffuse large B-cell lymphomaTumor tissues and cell linesGEO and WBPositively associated with favorable prognosis29Kidney chromophobeTumor tissuesTCGA and GEOPositively associated with favorable prognosis45CholangiocarcinomaTumor tissues and cell linesTCGA, GEO, IHC, and qRT-PCRPositively associated with favorable prognosis119,120Head and neck squamous cell carcinomaTumor tissuesGEO, TCGA, and IHCNegatively correlated with malignancy and T-cell exhaustion45Kidney renal papillary cell carcinomaTumor tissuesGEO, TCGA, and IHCPositively associated with favorable prognosis45Adrenocortical carcinomaTumor tissuesGEO, TCGA, and GTExAct as an independent prognostic predictor and is associated with poor prognosis, immune infiltration and immune checkpoints121Esophageal squamous cell carcinomaTumor tissuesGEO and TCGAAssociated with poor prognosis122FDX1: Ferredoxin 1; GEO: Gene Expression Omnibus; GTEx: Genotype-Tissue Expression Project; IHC: Immunohistochemistry; qRT-PCR: Quantitative reverse transcription-polymerase chain reaction; TCGA: The Cancer Genome Atlas; WB: Western blot.Figure 2**The regulatory mechanisms underlying aberrant FDX1 expression in cancer.**FDX1 expression is regulated by multiple transcription factors: YY1, NRF2, and FOXO3 act as transcriptional repressors, whereas SF-1 and NF-κB function as activators. FDX1 expression levels are inversely correlated with the methylation status of its promoter region across various cancer types. Lactylated NUDT21 suppresses FDX1 expression by promoting 3′ UTR elongation. The lncRNA PVT1 enhances FDX1 transcription through direct binding to its promoter region and facilitates the enrichment of H3K27ac, a histone modification associated with active enhancers. YTHDF1 and METTL16 positively regulate FDX1 expression by promoting m6A modification of its mRNA, while ALKBH5 negatively regulates FDX1 expression by reducing m6A levels. In addition, FDX1 expression is modulated by non-coding RNAs, including lncRNAs, circRNAs, and miRNAs. ALKBH5: AlkB homolog 5, RNA demethylase; circRNAs: circular RNAs; FDX1: Ferredoxin 1; FOXO3: Forkhead box O-3; lncRNAs: long non-coding RNAs; METTL16: Methyltransferase-like protein 16; miRNAs: microRNAs; NF-κB: Nuclear factor-κB; NRF2: Nuclear factor erythroid 2-related factor 2; NUDT21: Nudix hydrolase 21; SF-1: Steroidogenic factor-1; YTHDF1: YTH domain-containing family protein 1; YY1: Yin Yang 1; 3′-UTR: 3′-untranslated region.Figure 2
Transcription factors
Gene expression analyses from the Gene Expression Omnibus and The Cancer Genome Atlas databases show that FDX1 mRNA levels vary widely across various cancer types, indicating transcriptional regulation of FDX1 in cancer cells.44^,^45
Yin Yang 1 (YY1), a transcription factor belonging to the GLI-Kruppel family of zinc-finger proteins, plays a critical role in tumor initiation and progression.46 Wu et al. reported that SEC14-like lipid binding 3 induces cuproptosis by upregulating FDX1 expression through inhibition of the mitogen-activated protein kinase–extracellular signal-regulated kinase 1/2-YY1 signaling axis, revealing YY1 as a transcriptional repressor of FDX1.12 Nuclear factor erythroid 2-related factor 2 (NRF2), a transcription factor central to oxidative stress responses, also functions as a transcriptional inhibitor of FDX1 in cervical cancer cells.11 Additionally, phosphorylated forkhead box O3 (FOXO3) translocates to the nucleus and suppresses FDX1 transcription in ccRCC cells, establishing FOXO3 as another transcriptional repressor of FDX1.13 The collective inhibitory roles of YY1, NRF2, and FOXO3 suggest that combining a cuproptosis inducer with an inhibitors of these transcription factors may represent a promising strategy to enhance therapeutic efficacy. Conversely, FDX1 is transcriptionally upregulated in multiple cancer types. Steroidogenic factor-1 (SF-1) binds to the FDX1 promoter and enhances its transcriptional activity in colorectal cancer cells, a process facilitated by the long non-coding RNA (lncRNA) PVT1.7 Moreover, nuclear factor kappa B (NF-κB) acts as a positive transcriptional regulator of FDX1 by binding to its promoter. Knockdown of NF-κB significantly suppresses tumor growth, while co-overexpression of FDX1 partially reverses this effect in both in vitro and in vivo glioblastoma models.10 The transcriptional activation of FDX1 by SF-1 and NF-κB highlights a potential therapeutic strategy for cancers characterized by the activation of these factors. Overall, YY1, NRF2, and FOXO3 function as transcriptional repressors of FDX1, whereas SF-1 and NF-κB serve as transcriptional activators of FDX1 in cancer.
Epigenetic modifications
Aberrant epigenetic alterations can cause inappropriate upregulation or silencing of gene expression, contributing to cancer initiation and progression.47 Increasing evidence indicates that epigenetic modifications regulate FDX1 gene expression.
Across multiple cancer types, FDX1 gene expression shows a negative correlation with DNA methylation levels, suggesting that methylation contributes to FDX1 downregulation.15^,^48 By contrast, N6-methyladenosine (m6A) modification, the most prevalent RNA epigenetic modification, enhances FDX1 expression. The YTH N6-methyladenosine RNA-binding protein F1 (YTHDF1) promotes MYC-induced FDX1 expression in glioma cells by facilitating m6A modification of FDX1 mRNA,14 thereby supporting glioma cell proliferation and invasion through the MYC–YTHDF1–FDX1 axis.14 Moreover, copper stress induces lactylation of methyltransferase-like protein 16 (METTL16) at lysine 229, which upregulates FDX1 expression by enhancing the m6A modification of FDX1 mRNA in gastric cancer.16 Sirtuin 2 suppresses METTL16 lactylation; therefore, combining elesclomol, a copper ionophore, with AGK2, a sirtuin 2 inhibitor, enhances cuproptosis in gastric cancer.16 Histone acetylation also affects FDX1 expression. LncRNA PVT1 binds to the FDX1 promoter, increases acetylation of histone H3 at lysine 27, and activates FDX1 transcription.7 Furthermore, lactate dehydrogenase A-induced lactylation of nudix hydrolase 21 (NUDT21) extends the FDX1 mRNA 3′-untranslated region (3′-UTR) and represses its expression by interacting with cleavage and polyadenylation specificity factor 6 (CPSF6) in esophageal squamous cell carcinoma (ESCC).17 The combination of stiripentol, a lactate dehydrogenase A inhibitor, and elesclomol synergistically suppresses ESCC growth by targeting the NUDT21-CPSF6-FDX1 axis.17 Overall, epigenetic modifications play a key role in FDX1 dysregulation in cancer. The combination of cuproptosis inducers and epigenetic modulators may offer a promising therapeutic strategy for cancer treatment.
Non-coding RNAs
ncRNAs are single-stranded RNA molecules that play critical roles in regulating gene expression and contribute significantly to cancer progression.49 Major types of ncRNAs include microRNAs (miRNAs), lncRNAs, and circular RNAs (circRNAs). Increasing evidence shows that ncRNAs regulate FDX1 gene expression in various cancer types.
The oncogenic miRNA miR-21-5p directly binds to the 3′-UTR of FDX1 mRNA, promoting its degradation and thereby facilitating ccRCC progression.50 The miR-21-5p–FDX1 axis is also present in non-small cell lung cancer.23 Moreover, miR-3130-5p suppresses FDX1 expression by binding to its 3′-UTR in HCC cells.24 These findings indicate that miRNAs are key negative regulators of FDX1 expression in various cancers. By contrast, lncRNAs upregulate FDX1 expression by targeting miRNAs. Specifically, lncRNA 02362 enhances FDX1 by reducing miR-18-5p levels.19 Activation of the lncRNA 02362–miR-18-5p–FDX1 axis inhibits cell proliferation and increases oxaliplatin sensitivity through cuproptosis in HCC cells.19 Additionally, the lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) increases FDX1 expression by suppressing miR-328–3p in stomach adenocarcinoma.20 Conversely, knockdown of the lncRNA small nucleolar RNA host gene 7 (SNHG7) enhances FDX1 expression, which in turn inhibits proliferation, invasion, and metastasis by inducing cuproptosis in colorectal cancer.21 Beyond miRNAs and lncRNAs, circRNAs also regulate FDX1 expression. An 8-oxoguanine-modified circRNA derived from KIAA1797 binds to FDX1 mRNA, reducing its stability and suppressing cuproptosis, thereby promoting lung cancer progression.22 Collectively, these results identify FDX1 as a downstream target of ncRNAs in multiple cancer types. The ncRNA-FDX1 regulatory axis plays a critical role in cancer progression, making it a promising therapeutic target for cancer treatment.
Role of FDX1 in cancer progression
Accumulating evidence indicates that FDX1 dysregulation influences tumor development through multiple mechanisms, including modulation of oncogenic signaling pathways, metabolic reprogramming, and alterations in tumor immunity [Figure 3].Figure 3The underlying mechanisms by which FDX1 modulates cancer progression:signaling pathways, metabolic reprogramming, and tumor immunity. FDX1 plays a crucial role in cancer progression by modulating several key signaling pathways, including AMPK, mTOR, PI3K-AKT, IRF3-IFNβ, and Hippo-YAP. It is also essential for maintaining lipid homeostasis and regulating mitochondrial metabolism. Moreover, FDX1 expression is positively correlated with immune cell infiltration across various cancer types and is involved in the regulation of immune checkpoint genes, suggesting its potential involvement in antitumor immunity. Furthermore, FDX1 may contribute to remodeling the tumor immune microenvironment through the modulation of intratumoral metabolic processes. PI3K-AKT: hosphatidylinositol 3'-kinase(PI3K)-Protein kinase B(AKT); AMPK: AMP-activated protein kinase; FDX1: Ferredoxin 1; Hippo-YAP: Hippo-Yes-associated protein pathway; mTOR: Mechanistic target of rapamycin kinase; PI3K- IRF3-IFNβ: Interferon regulatory factor 3-interferon β.Figure 3
Signaling pathways
FDX1 is involved in several signaling pathways, including adenosine monophosphate-activated protein kinase (AMPK),28 mammalian target of rapamycin (mTOR),28 phosphatidylinositol 3′-kinase (PI3K)-AKT,5 IFN regulatory factor 3-interferon β (IRF3-IFNβ),29 and Hippo-Yes-associated protein (Hippo-YAP).30
AMPK and mTOR are essential for maintaining metabolic homeostasis and enabling metabolic adaptability.51 Liu et al. reported that FDX1 enhances autophagy by activating AMPK and inhibiting mTOR, thereby facilitating ovarian cancer progression.28 Compound C, a selective AMPK inhibitor, effectively suppresses tumor growth in ovarian cancer models with FDX1 overexpression.28 The PI3K-AKT pathway regulates tumor growth, drug resistance, invasion, and metastasis in various cancers.52 FDX1 knockdown activates this pathway by increasing reactive oxygen species (ROS) production, which enhances HCC growth and metastasis in vivo.5 By contrast, the PI3K-AKT pathway phosphorylates FDX1 to suppress its enzymatic activity in triple-negative breast cancer (TNBC).39 Combined treatment with AKT1 inhibitors and copper ionophores synergistically suppresses TNBC tumorigenesis in vitro and in vivo.39 These findings suggest a negative feedback loop between FDX1 and the PI3K-AKT pathway, highlighting a potential therapeutic target for cancer intervention. IFNβ, a cytokine of the type I interferon family, suppresses cancer progression by exerting direct antitumor effects and promoting immune responses.53 FDX1 has been demonstrated to activate the IFNβ signaling pathway by enhancing IRF3 phosphorylation, a key transcriptional activator of IFNβ, thereby inducing PANoptosis in diffuse large B-cell lymphoma cells following elesclomol treatment.29 These findings reveal an unrecognized functional role of FDX1 in elesclomol-induced cell death. The Hippo pathway functions as a tumor suppressor by regulating the activity of Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ) in a wide range of human cancers.54 FDX1 knockdown inhibits colorectal cancer progression by upregulating phosphorylated YAP levels and downregulating TAZ expression.30 The Hippo–YAP pathway inhibitor GA-017 effectively counteracts the inhibitory effects of FDX1 silencing on colorectal cancer progression.30 Overall, dysregulation of FDX1 influences the activity of multiple signaling pathways. Investigating the regulatory interactions between FDX1 and these pathways may enhance understanding of cancer progression mechanisms and support the development of potential anticancer strategies.
Metabolic reprogramming
FDX1 is involved in multiple metabolic pathways, particularly those associated with lipid homeostasis and mitochondrial metabolism. Disruptions of lipid homeostasis have been linked to various pathological conditions, including cancer.55 Suppression or depletion of ATP-binding cassette transporter A1 (ABCA1) leads to cholesterol accumulation, which subsequently activates sterol regulatory element-binding proteins 1 and 2 (SREBP1/2), key transcription factors that regulate lipid homeostasis in tumor cells.56 FDX1 deficiency promotes abnormal lipid accumulation through modulation of the ABCA1–SREBP1/2 signaling pathway.31 Furthermore, the FDXR–p53 axis plays an important role in lipid homeostasis and tumor progression.57 Loss of FDXR and/or p53 induces lipid accumulation in tumor cells via the downregulation of ABCA1 and activation of SREBP1/2.57 As a functional component of the FDXR complex, FDX1 may contribute to lipid homeostasis within the FDXR–p53 axis by modulating the ABCA1–SREBP1/2 pathway.
Mitochondrial metabolism plays a pivotal role in tumor progression by supplying essential metabolites for macromolecular synthesis and generating ATP to meet the heightened metabolic demands of cancer cells.58 FDX1 serves as a central regulator of mitochondrial metabolism, primarily through its role in protein lipoylation. Through direct interaction with LIAS, FDX1 promotes the assembly of the LIAS-GCSH complex, initiating lipoylation of target proteins.33 This post-translational modification enhances the enzymatic activity of four critical mitochondrial metabolic complexes: pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, branched-chain α-ketoacid dehydrogenase, and the glycine decarboxylase complex.33 These lipoylated complexes are integral to the TCA cycle, BCAA catabolism, and glycine metabolism. Cancer cells depend heavily on the TCA cycle for energy production and biosynthesis of cellular components.59 Therapeutic agents targeting this cycle have shown promising antitumor efficacy across multiple cancer types.60 BCAAs exhibit context-dependent roles in tumor progression, promoting proliferation by providing energy and biosynthetic intermediates in some cancers, while inducing cell cycle arrest or augmenting in immune-mediated tumor suppression others.61 Dysregulation of glycine cleavage metabolism also contributes to the progression of multiple cancers, including multiple myeloma62 and lung cancer,63 as demonstrated in both in vitro and in vivo studies. Given the critical role of these mitochondrial metabolic pathways, FDX1 may modulate cancer progression by regulating lipoylation-dependent metabolic processes.
FDX1 gene expression is closely linked to glucose metabolism, fatty acid oxidation, oxidative phosphorylation, and amino acid metabolism across multiple cancer types.64 In addition, FDX1 regulates key metabolic enzymes, such as glucose-6-phosphate dehydrogenase65 and hypoxia-inducible factor 1α.27 However, the precise mechanisms through which FDX1 modulates these metabolic pathways and enzyme activities remain unclear. Collectively, these results indicate that FDX1 plays a crucial role in the metabolic reprogramming of tumor cells. Further research is necessary to clarify the specific regulatory mechanisms and their contributions to tumor metabolism.
Tumor immunity
Despite significant advancements in immune checkpoint-based immunotherapy, only a subset of patients achieve durable clinical response, highlighting tumor immune evasion as a major obstacle to effective cancer treatment. Analysis of single-cell RNA sequencing data from 11 patients with glioma revealed that FDX1 is expressed in most immune cells within the tumor microenvironment. Glioma patients with high FDX1 expression also exhibit increased immune cell infiltration.34 In HCC tissues with elevated FDX1 expression, infiltration of natural killer (NK) cells, macrophages, and B cells notably increases.66 In ccRCC, FDX1 overexpression enhances antitumor immunity by promoting the secretion of interleukin-2 and tumor necrosis factor-γ.67 Conversely, impaired FDX1 enzymatic activity correlates with reduced immune cell infiltration in ccRCC.68 IFNβ has been shown to promote immune cell recruitment, including CD8^+^ T-cell and NK cell infiltration.69 These findings suggest that FDX1 may facilitate immune cell infiltration by activating the IRF3-IFNβ signaling pathway. YAP, a key component of the Hippo pathway, is positively and negatively correlated with M2 macrophage and regulatory T-cell infiltration in melanoma, respectively.70 This suggests that FDX1 may modulate immune cell infiltration through regulation of the Hippo–YAP pathway. Collectively, these findings imply that FDX1 contributes to enhanced immune cell infiltration within the tumor microenvironment.
FDX1 also regulates the expression of immune checkpoint genes. In ccRCC, FDX1 expression shows a positive correlation with several immune checkpoint genes, including programmed cell death ligand 1 (PD-L1), hepatitis A virus cellular receptor 2, and programmed cell death ligand 2. Conversely, FDX1 exhibits a negative correlation with cytotoxic T-lymphocyte-associated protein 4, lymphocyte activation gene 3, and programmed cell death 1 (PD-1).71 Glioma tissues with high FDX1 expression demonstrate elevated levels of immune checkpoint markers, such as PD-L1, CD40, CD80, and CD86.34 silencing FDX1 reduces PD-L1 expression in melanoma cells.72 Several oncogenic signaling pathways, including PI3K–AKT–mTOR and NF-κB, regulate immune checkpoint gene expression.73^,^74 These findings suggest that FDX1 may modulate immune checkpoint gene expression by regulating these signaling pathways.
Metabolism in both cancer and immune cells critically shapes the tumor immune microenvironment. In cancer cells, metabolic reprogramming enables the production and release of immunomodulatory molecules, such as ATP, mitochondrial DNA, high mobility group box 1, and α-ketoglutarate, which modulate antitumor immune responses.58^,^75^,^76 Mitochondrial metabolism-derived ROS also activate key transcription factors that support optimal T cell function.58 Memory T cells primarily depend on oxidative phosphorylation (OXPHOS) to support their metabolic needs.58 FDX1, expressed in both tumor and immune cells within the tumor microenvironment, plays a central role in regulating metabolism through protein lipoylation.34 Therefore, FDX1 may function as a metabolism-immune checkpoint gene through the dual regulation of metabolism in tumors and immune cells. Given its association with immune cell infiltration and immune checkpoint gene expression, FDX1 expression may serve as a predictive biomarker for immunotherapy response. Modulating FDX1 expression or activity could potentially enhance the efficacy of immune checkpoint inhibitors. Although correlations between FDX1 expression and features of the tumor immune microenvironment have been observed, the underlying molecular mechanisms remain unclear.
Molecular basis of FDX1's dual roles in cancer progression
Tumors undergo metabolic reprogramming that supports cancer progression.77 In mitochondrial metabolism-dependent cancers, FDX1 may promote tumor growth by enhancing mitochondrial enzyme activity through lipoylation. Meanwhile, FDX1 mediates cuproptosis and increases immune cell infiltration, potentially sensitizing tumors to cuproptosis inducers and immunotherapy. In cancers that rely on aerobic glycolysis, FDX1 overexpression may suppress cancer progression by shifting cellular metabolism from aerobic glycolysis to OXPHOS. Additionally, cancer cells reprogram their metabolism to support their survival during tumor progression. In the early stages, aerobic glycolysis supports proliferation by providing energy and biosynthetic intermediates,78 whereas in advanced stages, OXPHOS contributes to drug resistance, radioresistance, and metastasis.79 Single-cell sequencing data reveal that FDX1 expression increases from early to advanced stages in several cancer types,44^,^45 suggesting that FDX1 regulates this metabolic transition. Through the activation of OXPHOS to support energy supply for metastasis, FDX1 may promote tumor progression in advanced cancers while simultaneously increasing sensitivity to cuproptosis inducers. The mitochondrial metabolic profiles of cancer may therefore determine the functional role of FDX1 during cancer progression.
Autophagy suppresses tumorigenesis by limiting cancer cell survival; however, it can also promote resistance to chemotherapy and radiotherapy while facilitating tumor metastasis.80 Therefore, FDX1 may inhibit tumorigenesis yet promote tumor progression by inducing autophagy. Given that FDX1 serves as a key mediator of cuproptosis, copper levels within the tumor microenvironment may modulate its functional role in cancer progression. The liver, the primary organ for copper storage in the human body,81 enables FDX1 to function as a tumor suppressor in HCC by mediating cuproptosis. By contrast, the Hippo–YAP signaling pathway acts as a tumor suppressor in colorectal cancer,82 but promotes tumor progression in breast cancer,83 which may explain FDX1's tumor-promoting role in colorectal cancer and tumor-inhibiting function in breast cancer. These signaling pathways, therefore, contribute to the dual functions of FDX1 in cancer progression. Collectively, cancer type, metabolic characteristics, autophagy, copper levels in the tumor microenvironment, signaling pathways, and cuproptosis may contribute to the dual roles of FDX1 in cancer progression.
Therapeutic potential of FDX1
FDX1 plays a crucial role in the progression of multiple cancers and is a key mediator of cuproptosis. Targeting FDX1 is a promising therapeutic strategy for cancer treatment. Several recent approaches have been developed to directly or indirectly modulate FDX1 activity, demonstrating its therapeutic potential in in vitro and in vivo cancer models.
Copper ionophores
The elesclomol-Cu^2+^ complex to directly bind to the α2/α3 helices and β5 strand of FDX1, with no detectable interaction with its paralog FDX2.84 This binding induces complete oxidation of FDX1 and promotes Cu ^+^ generation, ultimately triggering cuproptosis. Substantial evidence indicates that elesclomol exerts anticancer effects through FDX1-mediated cuproptosis in various cancer types.85, 86, 87, 88 Disulfiram, another copper ionophore initially approved for alcohol aversion therapy, also exhibits promising anticancer activity by enhancing FDX1-mediated cuproptosis.89 The therapeutic efficacy of copper ionophores, either as monotherapy or in combination with other agents, has recently been evaluated in multiple cancer trials across different cancer types [Table 2]. However, most clinical trials have not achieved the desired clinical responses. Limited efficacy, safety concerns, and pharmacokinetic limitations contribute to these shortcomings.90 Both copper ionophores undergo rapid plasma clearance and exhibit short half-lives, making it difficult to maintain sustained therapeutic concentrations.90 Safety issues, such as hepatic copper accumulation, cardiotoxicity, and neurotoxicity, were major factors in the failure of the elesclomol clinical trial.90^,^91 Nanoparticle-based delivery systems for copper ionophores may improve selectivity and enhance accumulation within tumor tissues.26^,^92^,^93 Patients with melanoma and low lactate dehydrogenase (LDH) levels exhibit better responses to elesclomol than those with high LDH levels.94 Elevated tumor LDH is associated with hypoxia and reflects a glycolytic phenotype in cancer cells, supporting the therapeutic potential of copper ionophores in metabolic targeting strategies. Furthermore, combining copper ionophores with other anticancer agents that synergize with their mechanisms of action may increase their effectiveness. Several studies have found that copper ionophores exhibit increased potency when combined with glycolysis inhibitors,95 platinum compounds,88 or proteasome inhibitors.85Table 2. The clinical trials of copper ionophores.Table 2. Cuproptosis related drugsClinical trial phaseStudy populationMain resultsClinical trials IDReferencesElesclomol in combination with paclitaxelIIIMetastatic melanoma patientsEffective in a subset of patients (LDH potentially a predictive marker for the combination regimen's suitability).NCT0052283494Elesclomol sodium in combination with paclitaxelIIPatients with metastatic melanoma, measurable disease, and one or fewer prior chemotherapyElesclomol sodium combined with paclitaxel significantly doubled median PFS, with acceptable toxicity and promising OS.NCT00084214123Elesclomol sodiumIAcute myeloid leukemia patientsElesclomol showed good safety (max dose 400 mg/m^2^, no dose-limited toxicity) but no clinical responses in AML patients.NCT01280786124Elesclomol sodium in combination with paclitaxelIIPatients with platinum-resistant recurrent ovarian, tubal or peritoneal cancerThis combination was well tolerated but unworthy of further investigation due to response proportion.NCT00888615125Elesclomol sodium in combination with paclitaxelIAdults with refractory solid tumorsThe elesclomol/paclitaxel combination was well tolerated, with enhanced paclitaxel exposure (dose- dependently interacting with elesclomol) causing increased toxicity; responses in two pretreated patients support further evaluation.NCT00088114126Elesclomol sodium in combination with taxaneIISoft tissue sarcomaElesclomol enhanced taxane efficacy by induction of HSP70.NCT00087997127Elesclomol sodium plus paclitaxel, carboplatinI/IIStage IIIB/IV non-small cell lung cancerUnpublishedNCT00088088N/AElesclomol sodium plus docetaxel and prednisoneIPatients with metastatic prostate cancerUnpublishedNCT00808418N/ADisulfiram/CuIPatients with advanced solid tumors involving the liverDisulfiram combined with copper gluconate was well-tolerated and safe in liver solid tumor patients. It led to temporary disease stabilization in some but no objective responses.NCT00742911128Disulfiram/CuIIMetastatic breast cancer patients upon failure of conventional systemic and/or locoregional therapiesUnpublishedNCT03323346N/ADisulfiram/Cu in combination with cisplatin and vinorelbineIICTC_EMT positive refractory metastatic hormone receptor positive, HER2 negative breast cancer patientsUnpublishedNCT04265274N/ADisulfiram and cisplatinIIPatients with multiple relapsed/refractory GCTsUnpublishedNCT03950830N/ADisulfiramNARecurrent prostate cancer patientsUnpublishedNCT01118741N/ADisulfiram/CuIbPancreas cancer patients metastatic castrate-resistant prostate cancer patientsUnpublishedNCT02963051N/ADisulfiram in combination with cisplatin and vinorelbineIIbPatients with non-small cell lung cancerThe addition of disulfiram to a combination regimen of cisplatin and vinorelbine was well tolerated and appeared to prolong survival in patients with newly diagnosed non-small cell lung cancer.NCT00312819129Disulfiram and cisplatinNAPatients with advanced gastric cancerUnpublishedNCT05667415N/ADisulfiram in combination with chemotherapyIPatients with refractory solid tumors or metastatic pancreatic cancerUnpublishedNCT02671890N/ADisulfiram and chelated zincIIRefractory disseminated malignant melanoma patientsUnpublishedNCT02101008N/ADisulfiram Plus arsenic TrioxideIMetastatic melanoma patientsUnpublishedNCT00571116N/ADisulfiramI/IIMetastatic melanoma patientsUnpublishedNCT00256230N/ADisulfiram and metforminI/IISuspected glioblastoma or recurrent glioblastoma patients undergoing surgical resectionUnpublishedNCT03151772N/ADisulfiram/Cu with concurrent radiation therapy and TMZI/IIPatients with newly diagnosed glioblastomaThe combination regimen has limited clinical efficacy for most patients but shows promising efficacy in BRAF-mutant glioblastoma.NCT02715609130Disulfiram/Cu in combination with TMZIIRecurrent TMZ-resistant glioblastoma patientsAdding disulfiram/Cu to TMZ for TMZ-resistant IDH-wild type glioblastoma is well tolerated, yet shows limited activity in an unselected population.NCT03034135131Disulfiram/CuIINewly diagnosed glioblastoma patientsUnpublishedNCT01777919N/ADisulfiram in combination with alkylating agentsIPatients with glioblastomaUnpublishedNCT01907165N/ADisulfiram in combination with temozolomideIIUnmethylated glioblastoma patientsUnpublishedNCT03363659N/ADisulfiram/Cu in combination with alkylating chemotherapyII/IIIPatients with recurrent glioblastomaFor patients with recurrent glioblastoma, adding disulfiram and copper to chemotherapy increased toxic effects without improving survival.NCT02678975132Disulfiram/Cu in combination with liposomal doxorubicinIPatients with treatment-refractory sarcomasUnpublishedNCT05210374N/ADisulfiram/CuIPatients with treatment-refractory Multiple MyelomaUnpublishedNCT04521335N/ADisulfiram/CuIIMetastatic pancreatic cancer patientsUnpublishedNCT03714555N/AAML: Acute myeloid leukemia; BRAF: B-raf proto-oncogene, serine/threonine kinase; GCTs: Germ cell tumors; HER2: Erb-B2 receptor tyrosine kinase 2; HSP70: Heat shock protein 70; IDH: Isocitrate dehydrogenase; LDH: Lactate dehydrogenase; N/A: Not available; OS: Overall survival; PFS: Progression free survival; TMZ: Temozolomide.
Small-molecule-mediated upregulation or activation of FDX1
Given the essential role of FDX1 in cuproptosis, increasing FDX1 levels or activity may provide an effective strategy for enhancing the sensitivity of cancer cells to cuproptosis inducers. The ferroptosis inducers sorafenib and erastin upregulate protein lipoylation by suppressing the mitochondrial matrix protease-mediated degradation of FDX1 and reducing intracellular glutathione synthesis by inhibiting cystine import, thereby inducing cuproptosis in liver cancer cells.38 Combining the elesclomol-Cu^2+^ complex with sorafenib or erastin synergistically suppresses liver cancer progression in xenograft mouse models by enhancing cuproptosis.38 [1-propyl-3,5-bis(2-bromobenzylidene)-4-piperidinone], a novel curcuminoid, effectively inhibits the proliferation, invasion, and migration of cervical cancer cells.11 Mechanistic studies have demonstrated that this curcuminoid upregulates FDX1 expression by inhibiting the neurogenic locus notch homolog protein 1 (Notch1)–recombination signal-binding protein for the immunoglobulin kappa J region (RBP-J)–NRF2 signaling pathway, leading to cuproptosis in cervical cancer cells.11 In TNBC, AKT1 phosphorylates FDX1 protein and inhibits its activity, thereby inducing cuproptosis resistance.39 The combination of MK2206, a selective AKT1 inhibitor, with the elesclomol-Cu^2+^ complex synergistically suppresses TNBC tumorigenesis by enhancing cuproptosis in vitro and in vivo.39 Fluphenazine, a first-generation antipsychotic, induces cuproptosis by upregulating FDX1 expression at both the mRNA and protein levels in breast cancer.40 Collectively, these findings suggest that enhancing FDX1 expression or augmenting its protein activity is a promising therapeutic strategy.
Combination immunotherapy
FDX1 expression positively correlates with immune cell infiltration in several cancer types. Moreover, FDX1 regulates the expression of immune checkpoint genes,34^,^35^,^66^,^72 making it a potential predictor of the immunotherapy response in cancers, including ccRCC,67 HCC,66 glioma,34 and colon adenocarcinoma.96 Upregulation or activation of FDX1 may facilitate the conversion of “cold tumors” into “hot tumors,” which respond more effectively to immunotherapy. High FDX1 expression has been associated with improved responses to PD-L1 blockade in melanoma.72 Drug sensitivity analyses revealed that FDX1 expression is positively correlated with the response to PD-1 inhibitor therapy in patients with melanoma and kidney cancers.97 Combining an FDX1 activator with an immune checkpoint inhibitor may therefore offer a promising therapeutic approach. Recent studies demonstrate that copper ionophores combined with immune checkpoint inhibitors, such as anti-CD47 antibody, JQ-1, and anti-PD-1 (aPD1), yield significant therapeutic potential in multiple cancers.93^,^98^,^99 Given that FDX1 functions as a central regulator of both cuproptosis and the tumor immune microenvironment, it may act as an essential mediator of the synergistic effects observed between copper ionophores and immunotherapy checkpoint inhibitors.
Other therapeutic strategies
Upregulation of FDX1 expression has been observed in various forms of drug resistance and radioresistance across multiple cancer types,15 highlighting the therapeutic potential of cuproptosis in these malignancies. Under severe hypoxia, FDX1 expression increases and promotes radioresistance in glioblastoma.27 Radiotherapy elevates mitochondrial copper levels by upregulating copper transporter 1 expression and reducing mitochondrial glutathione, thereby triggering cuproptosis in patient-derived xenografts.100 These findings indicate that radiotherapy may synergize with copper ionophores to overcome tumor radioresistance through FDX1-mediated cuproptosis. Moreover, the observed upregulation of FDX1 in tumors following radiotherapy supports the use of copper-containing nanocapsule-like polyoxometalates, which have demonstrated efficacy in overcoming acquired radioresistance, even at clinically relevant radiation doses, in preclinical tumor models.26 FDX1-mediated cuproptosis has been shown to overcome various forms of drug resistance in tumor cells and preclinical models, such as enzalutamide resistance in prostate cancer,86 cisplatin resistance in ovarian cancer,25 adrenomedullin-mediated sunitinib resistance in ccRCC,13 and lenvatinib resistance in HCC.101 Therefore, FDX1-mediated cuproptosis represents a promising therapeutic strategy for addressing both drug resistance and radioresistance in cancer.
Crosstalk among regulated cell death modalities involves multiple key mediators,102 and targeted modulation of these mediators may provide promising therapeutic strategies for cancer treatment.103 In addition to cuproptosis, excessive copper induces other forms of cell death, including apoptosis, pyroptosis, and ferroptosis.104 Copper-induced ROS generation and proteotoxicity constitute the primary mechanisms underlying copper-mediated cytotoxicity. FDX1 reduces Cu^2+^ to Cu^+^, which subsequently enhances ROS generation via the Fenton reaction and leads to aggregation of lipoylated proteins. Therefore, FDX1 may play a pivotal role in mediating copper-induced cell death. A core–shell nanoparticle incorporating copper and erastin, a ferroptosis inducer, has demonstrated significant anticancer efficacy by synergistically activating both cuproptosis and ferroptosis in murine models of colon adenocarcinoma and TNBC.105 Similarly, a copper-based metal–organic framework (Cu-MOF) exhibits enhanced antitumor activity in vitro and in vivo through the co-activation of apoptosis and cuproptosis.106 These findings highlight the potential of combining cuproptosis with other regulated cell death pathways through engineered combination therapy nanoparticles.
Aggregation of lipoylated proteins is the primary mechanism underlying cuproptosis; therefore, upregulation of protein lipoylation-related genes, such as LIAS, dihydrolipoamide S-acetyltransferase (DLAT), GCSH, dihydrolipoamide s-succinyltransferase, and dihydrolipoamide branched-chain transacylase E2, may enhance cuproptosis in cancer. FDX1 regulates cellular protein lipoylation by directly binding to LIAS. Elevated LIAS expression in multiple cancers increases lipoylated protein levels, thereby enhancing susceptibility to cuproptosis. Deletion of FDX1 or LIAS confers resistance to cuproptosis.107 DLAT is overexpressed and serves as a promising prognostic marker and therapeutic target in most cancers.108 FDX1-mediated accumulation of lipoylated DLAT aggregation is a key mechanism underlying cuproptosis in cancer cells. Given the critical roles of these genes, a gene signature comprising FDX1 and other cuproptosis-related genes may serve as a biomarker for identifying cancers susceptible to cuproptosis.
Conclusions and future perspectives
Overall, FDX1 expression is dysregulated across various cancer types. Its expression is significantly associated with prognosis, disease progression, cuproptosis, and tumor immunity in multiple malignancies, highlighting its potential as a biomarker for cancer diagnosis, prognostic evaluation, and treatment decision-making. Several factors, including cancer type, metabolic characteristics, autophagy, copper levels within the tumor microenvironment, signaling pathways, and cuproptosis, may contribute to FDX1's dual roles in different cancers. Therapeutic strategies targeting FDX1 directly or indirectly have demonstrated promising efficacy in multiple cancer models.
Enhanced expression or activity of FDX1 produces significant synergistic effects when combined with copper ionophores in various malignancies. Moreover, FDX1 influences cancer progression by modulating signaling pathways, metabolic reprogramming, and tumor immune responses. Dynamic modeling of signaling networks has elucidated key regulatory mechanisms and enabled the systematic identification of potential therapeutic targets.109 Understanding the upstream and downstream regulatory mechanisms of FDX1 may reveal novel strategies to enhance its expression or functional activity, thereby facilitating the development of innovative therapeutic approaches for cancer treatment. FDX1 is expressed in tumor cells, immune cells, and fibroblasts; thus, single-cell RNA sequencing could elucidate its distinct functional roles within the tumor microenvironment.
Copper ionophores such as elesclomol and disulfiram demonstrate substantial anticancer activity in multiple preclinical models; however, their clinical efficacy in human trials remains limited. Virtual screening approaches based on the crystal structure of the FDX1-Cu^2+^ complex may aid in designing novel copper ionophores with improved translational potential for cuproptosis-targeted therapies. Patient-derived xenograft models have been widely used in preclinical research38^,^100 and provide clinically relevant platforms for advancing the development of cuproptosis inducers. Furthermore, combining copper ionophores with other anticancer agents may yield synergistic therapeutic effects. Collectively, further research is warranted to elucidate the mechanisms by which FDX1 modulates cancer progression and to clarify its functional implications.
Limitation of the review
This review summarizes the regulatory mechanisms, functional roles, and therapeutic potential of FDX1 in cancer; however, several limitations must be acknowledged. First, the dysregulation of FDX1 and its association with cancer prognosis are primarily based on data from the TCGA and GEO databases. The absence of protein-level validation compromises the reliability of the conclusions. Second, the clinical responses to copper ionophores in clinical trials remain limited, with most studies still relying on in vitro or animal models. The lack of efficacy, safety concerns, and pharmacokinetic limitations may contribute to the limited clinical response to copper ionophores. Third, the dual roles of FDX1 in different cancer types reduce the therapeutic value of targeting FDX1. Therefore, it is imperative to clarify the functional role of FDX1 in cancer and to develop both activators and inhibitors of FDX1. Addressing these challenges through immunohistochemical staining, improved delivery systems, combination therapy strategies, and the rational design of novel FDX1-targeting agents is crucial for advancing the clinical application of FDX1-targeted therapies.
Authors contribution
Jiliang Xia, Fen He, and Yongyan Zhao wrote the manuscript; Ruixin Gao, Maoyou Lu, Siqi Wang, and Yu Chen created the figures and table; Jiliang Xia and Lijun Peng revised the manuscript. All authors have read and approved the final version of the manuscript.
Data availability statement
The supporting data for this paper comes from published materials that are properly referenced and are available online.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors declare that generative artificial intelligence (AI) and AI assisted technologies were not used in the writing process or any other process during the preparation of this manuscript.
Funding
This work was supported by the 10.13039/501100004735Natural Science Foundation of Hunan Province (No.2023JJ10036), the PhD Scientific Research Start-up Fund of the 10.13039/501100009020University of South China (No.200XQD075), the Scientific Research Project of Hunan Department of Education (No.22B0452), the Innovation Platform Open Fund Project of Department of Education of Hunan Province (No.20K110).
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sheftel A.D.Stehling O.Pierik A.J.Humans possess two mitochondrial ferredoxins, Fdx 1 and Fdx 2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis Proc Natl Acad Sci U S A 1072010117751178010.1073/pnas.100425010720547883 PMC 2900682 · doi ↗ · pubmed ↗
- 2Shi Y.Ghosh M.Kovtunovych G.Crooks D.R.Rouault T.A.Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis Biochim Biophys Acta 1823201248449210.1016/j.bbamcr.2011.11.00222101253 PMC 3546607 · doi ↗ · pubmed ↗
- 3Cai K.Tonelli M.Frederick R.O.Markley J.L.Human mitochondrial ferredoxin 1 (FDX 1) and ferredoxin 2 (FDX 2) both bind cysteine desulfurase and donate electrons for iron-sulfur cluster biosynthesis Biochemistry 56201748749910.1021/acs.biochem.6b 0044728001042 PMC 5267338 · doi ↗ · pubmed ↗
- 4Zhao Q.Yu M.Du X.The role of cuproptosis key factor FDX 1 in gastric cancer Curr Pharm Biotechnol 26202413214210.2174/011389201030199724052716242338918976 · doi ↗ · pubmed ↗
- 5Sun B.Ding P.Song Y.FDX 1 downregulation activates mitophagy and the PI 3K/AKT signaling pathway to promote hepatocellular carcinoma progression by inducing ROS production Redox Biol 75202410330210.1016/j.redox.2024.103302 PMC 1136691339128228 · doi ↗ · pubmed ↗
- 6Chen J.Cao W.Li Y.Zhu J.Comprehensive analysis of the expression level, prognostic value, and immune infiltration of cuproptosis-related genes in human breast cancer Medicine (Baltim)1032024 e 4013210.1097/MD.0000000000040132 PMC 1149572539432636 · doi ↗ · pubmed ↗
- 7Ma J.Zhang Y.Sun Z.Lnc RNA PVT 1 promotes cuproptosis through transcriptional activation of FDX 1 in colorectal cancer Redox Biol 85202510372210.1016/j.redox.2025.103722 PMC 1220556240505346 · doi ↗ · pubmed ↗
- 8Yang W.Wu C.Jiang C.FDX 1 overexpression inhibits the growth and metastasis of clear cell renal cell carcinoma by upregulating FMR 1 expression Cell Death Discov 11202511510.1038/s 41420-025-02380-540118855 PMC 11928736 · doi ↗ · pubmed ↗