The complete chloroplast genome of Camellia melliana (Theaceae)
Mengyuan Xu, Quannian Li, Haiguang Gong, Shaoshan Luo, Jiuxiang Huang

TL;DR
This paper reports the complete chloroplast genome of the endangered Camellia melliana, providing insights into its genetic structure and phylogenetic relationships within the Camellia genus.
Contribution
The study presents the first complete chloroplast genome sequence of Camellia melliana and establishes its phylogenetic position within the genus.
Findings
The chloroplast genome of Camellia melliana is 156,984 bp in length and contains 112 unique genes.
Phylogenetic analysis shows a close relationship between C. melliana and C. salicifolia.
The study provides a foundation for future research on Camellia phylogenetics and genetic diversity.
Abstract
Camellia melliana Hand. -Mazz. is an endangered shrub species endemic to China, but it has not been sequenced and has never been included in molecular phylogenetic studies to date. In the present study, the complete chloroplast genome sequence of the species was assembled through the genome-skimming approach, and the phylogenetic position of the species within Camellia was investigated for the first time. Results showed that the chloroplast genome of the species is 156,984 bp in length, including a large single copy (LSC) region of 86,588 bp and a small single-copy (SSC) region of 18,268 bp, which were separated by a pair of inverted repeat (IR) regions of 26,064 bp. The genome encoded 112 unique genes, including 79 protein-coding genes, four ribosomal RNA genes and 29 transfer RNA genes. The overall GC content of the complete genome is 37.3%. Results from phylogenetic analysis…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1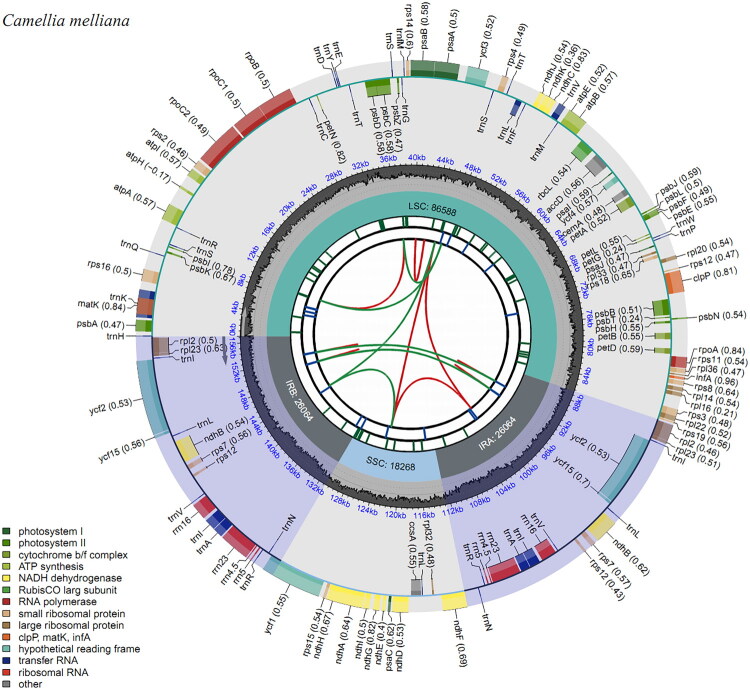 Figure 2
Figure 2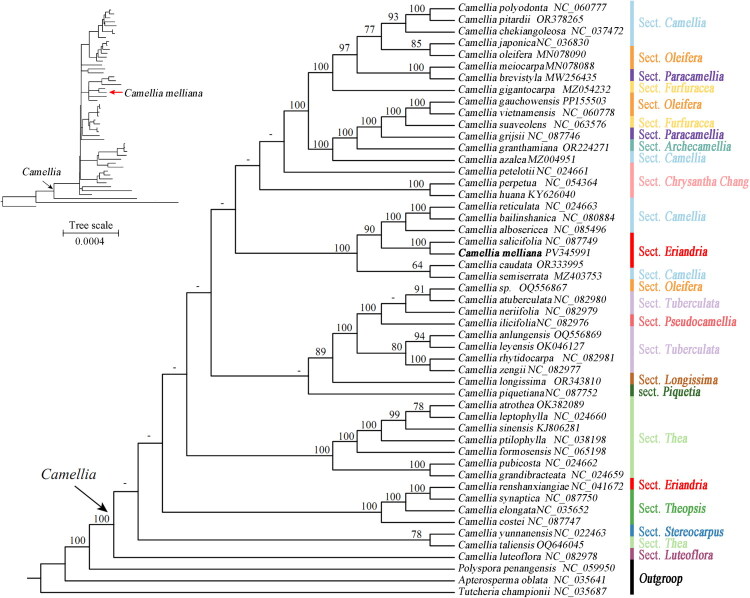 Figure 3
Figure 3- —Forestry Science and Technology Innovation Project of Guangdong Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant and Fungal Species Descriptions · Plant Diversity and Evolution · Genomics and Phylogenetic Studies
Introduction
Camellia L., comprising approximately 232 tree and shrub species, represents the largest genus within the tea family Theaceae (POWO 2025). Over 80% of its species diversity is concentrated in China (Chang and Ren 1998). According to Chang’s taxonomic system, Chinese Camellia species have been classified into four subgenera and 18 sections based on comprehensive morphological analyses of floral and fruit characteristics (Chang and Ren 1998). The genus holds significant economic and ornamental importance, with numerous species utilized for tea production, oil extraction, and horticultural purposes (Wu et al. 2022).
Camellia melliana Hand.-Mazz. 1922, an evergreen shrub endemic to Guangdong Province, China, belongs to section Eriandria Coh. St. according to Chang’s taxonomic system (Chang and Ren 1998). The species was listed as endangered in the Redlist of China’s Biodiversity (MEE 2023), while it represents an important component in evergreen broad-leaved forests in Guangdong Province (Zou et al. 2014). Although phylogenetic studies focusing on the large genus Camellia have been extensively conducted in recent years, C. melliana has remained unsequenced, and no DNA sequence data for this species have been reported to date. In the present study, we sequenced and analyzed the complete chloroplast genome of C. melliana and investigated its phylogenetic position within Camellia for the first time.
Materials and methods
Plant material of Camellia melliana was collected from the type locality of the species, viz. Chenhedong Provincial Nature Reserve in Guangzhou, Guangdong Province, China (N23°44′41.55″ E113°55′47.36″). The voucher specimen (Shaoshan Luo CHD20230812; Figure 1) was deposited in the Herbarium of South China Agricultural University (CANT; Index Herbariorum: https://sweetgum.nybg.org/science/ih/herbarium-details/?irn=126001; Curator: Prof. Yongbin Wu, email: [email protected]). Total genomic DNA was extracted from approximately 10 mg silica gel-dried leaf tissue using a modified CTAB protocol (Doyle and Doyle 1987). The chloroplast genome of C. melliana was sequenced through genome-skimming following the methodology of Xue et al. (2024). DNA was sheared to short fragments through ultrasonic treatment, and then the fragments approximately 500-bp in length were selected and used to construct short-insert library following the manufacturer’s protocol (NEBNext ® Ultra II ™DNA Library Prep Kit for Illumina®). Paired-end sequencing (2 × 150 bp) was performed on the Illumina HiSeq 2500 platform at Beijing Genomics Institute (BGI, Shenzhen, China), yielding approximately 3 GB of raw data. De novo assembly was conducted with GetOrganelle (Jin et al. 2020) using the chloroplast genome of Camellia caudata Wall. (OR333995) as a reference, which was chosen as a high-quality plastome from the same genus to facilitate accurate chloroplast read recruitment and assembly, given its completeness and annotation quality. Gene annotation was performed with the Plastid Genome Annotator (Qu et al. 2019). The complete chloroplast genome sequence has been deposited in NCBI GenBank (accession: PV345991; https://www.ncbi.nlm.nih.gov). A physical genome map was generated using CPGView (Liu et al. 2023; Figure 2).
(A) The voucher specimen of Camellia melliana, collected from Chenhedong Provincial Nature Reserve in Guangzhou, Guangdong Province, China (prepared by Shaoshan Luo). Photographs of Camellia melliana taken by Yousheng Chen in Yangjiang City, Guangdong Province (B and C). Permission to use the photographs was obtained from Yousheng Chen. Diagnostic features of Camellia melliana include pubescent young branches, oblong-lanceolate leaves (3–5 cm long) with a bluntly apiculate apex, and white axillary flowers with 5–6 petals partially adnate to the stamens.
Schematic map of overall features of the chloroplast genome of Camellia melliana. The map contains six tracks in default. From the Center outward, the first track shows the dispersed repeats. The dispersed repeats consist of direct (D) and palindromic (P) repeats, connected with red and green arcs. The second track shows the long tandem repeats as short blue bars. The third track shows the short tandem repeats or microsatellite sequences as short bars with different colors. The small single-copy (SSC), inverted repeat (IRa and IRb), and large single-copy (LSC) regions are shown on the fourth track. The GC content along the genome is plotted on the fifth track. The genes are shown on the sixth track. The optional codon usage bias is displayed in the parenthesis after the gene name. Genes are color-coded by their functional classification which is shown in the bottom left corner. The transcription directions for the inner and outer genes are clockwise and anticlockwise, respectively.
To investigate the phylogenetic position of Camellia melliana, complete chloroplast genome sequences from 47 additional Camellia species were retrieved from the NCBI GenBank database (Supplementary material, Table 1). Three taxa from closely related genera, Apterosperma H.T. Chang, Polyspora Sweet and Tutcheria Dunn, were selected as outgroups based on the phylogenetic framework of Theaceae proposed by Yu et al. (2017). Eighty-three coding regions, including 79 protein-coding genes and four ribosomal RNA (rRNA) genes (Supplementary material, Table 2) in plastome were extracted and then aligned using the MAFFT algorithm (Katoh et al. 2019). The aligned gene matrices were concatenated and used to reconstruct the phylogenetic tree. Detailed information regarding these genes and their lengths are provided in Supplementary material, Table 2. The maximum likelihood (ML) approach implemented in RAxML version 8.1.24 (Stamatakis 2006) was used to infer the phylogenetic tree, employing the GTRGAMMA model with the default number of rate categories (C = 25). The model accounts for among-site rate heterogeneity and is widely used for robust plastome-scale ML phylogenetic inference. A rapid bootstrap (BS) analysis with 1000 pseudoreplicates was conducted to obtain support values for each phylogenetic node.
Results
Structural analysis of the complete chloroplast genome of Camellia melliana revealed a typical quadripartite circular structure with 156,984 bp in length (Figure 2). The plastome exhibited an average read mapping depth of approximately 281× (Supplementary material, Figure 1). The final plastome assembly was gap-free (no ambiguous bases) and circularized. The genome comprises four distinct regions: a large single-copy (LSC) region of 86,588 bp, a small single-copy (SSC) region of 18,268 bp, and a pair of inverted repeat regions (IRa and IRb; 26,064 bp each). A total of 112 unique genes were annotated, including 79 protein-coding genes, four ribosomal RNA (rRNA) genes, and 29 transfer RNA (tRNA) genes. Several genes contained introns, including cis-splicing genes (e.g. rpoC1, ycf3 and clpP) and the trans-splicing gene rps12, whose structures are shown in Supplementary material, Figure 2. Duplicated genes within the IR regions include seven protein-coding genes (ndhB, rpl2, rpl23, rps12, rps7, ycf15, ycf2), four rRNA genes (rrn4.5, rrn5, rrn16, rrn23), and seven tRNA genes (trnA-UGC, trnL-CAA, trnI-CAU, trnI-GAU, trnN-GUU, trnR-ACG, trnV-GAC). The overall GC content of the C. melliana chloroplast genome was calculated as 37.3%.
The phylogenetic analysis strongly supported the monophyly of the genus Camellia (BS = 100%) and revealed a well-supported sister relationship between the genus and Polyspora (BS = 100%; Figure 3). Notably, the species C. melliana formed a strongly supported sister clade (BS = 100%) with C. salicifolia Champ. ex Benth (Figure 3). Camellia salicifolia has been placed in sect. Eriandria in morphology-based classifications (Chang and Ren 1998) and the recovered relationship is therefore congruent with the sectional placement of C. melliana. This congruence also aligns with the general utility of plastome phylogenomics for resolving relationships among closely related Camellia taxa reported in previous studies. In addition, several major clades within Camellia were resolved with high statistical support, but the majority of backbone nodes in the genus exhibited weak phylogenetic signals and resolved with weak support. This topological ambiguity suggests that incorporating additional molecular markers, particularly nuclear genomic data, would be essential for elucidating evolutionary relationships among major lineages in this species-rich genus.
The maximum likelihood (ML) tree of sampled species of Theaceae based on analysis of 83 coding regions of chloroplast genomes. ML bootstrap percentages over 50% are given near the nodes, with dashes denoting a support inferior to 50%. Bold type marks species sequenced in the present study. Section assignments of Camellia species follow Chang and Ren (1998) and are indicated on the right of the cladogram. An inset phylogram with branch lengths proportional to substitutions per site (tree scale shown) is provided in the upper-left to complement the topology-focused main tree. Numbers following the species names represent GenBank accession numbers and the corresponding publications are as follows:Camellia atrothea (OK382089) (Wang et al. 2023); Camellia azalea (MZ004951) (Xu et al. 2023); Camellia brevistyla (MW256435) (Yin et al. 2021); Camellia gigantocarpa (MZ054232) (Xu et al. 2022); Camellia ptilophylla (NC_038198) (Li et al. 2018); Camellia semiserrata (MZ403753) (Dong et al. 2021); Camellia anlungensis (OQ556869), Camellia leyensis (OK046127) (Ran et al. 2024); Camellia huana (KY626040) (Wang et al. 2017); Camellia leptophylla (NC_024660), Camellia petelotii (NC_024661), Camellia pubicosta (NC_024662), Camellia reticulata (NC_024663), Camellia synaptica (NC_087750) and Camellia taliensis (OQ640645) (Huang et al. 2014); Camellia sinensis (KJ806281) and Camellia yunnanensis (NC_022463) (Yang et al. 2013); Camellia granthamiana (OR224271) (Chen et al. 2023). In addition, the following sequences were used as outgroups:Polyspora penangensis (NC_059950) (Choo et al. 2020), Apterosperma oblata (NC_035641) and Tutcheria championii (NC_035687) (Yu et al. 2017).
Discussion and conclusion
Beyond phylogenetic inference, the newly generated plastome sequence also provides baseline genomic information for conservation-oriented assessments of threatened Camellia lineages. Given that several species in sect. Eriandria have been assessed as threatened in national red-list assessments, such genomic baseline resources may facilitate evidence-based conservation prioritization by improving taxonomic resolution and enabling downstream population genetic analyses. Accordingly, our data may serve as a reference for future reassessments and potential updates of protection priorities for threatened taxa.
In the present study, the endangered Chinese endemic species Camellia melliana was sequenced for the first time and its complete chloroplast genome sequence was provided. The phylogenetic position of the species within the large genus Camellia was also investigated here for the first time using molecular phylogenetic analysis, and a highly supported sister relationship between the species and C. salicifolia was recovered. This newly sequenced chloroplast genome provides valuable genomic data for advancing both conservation genetics and taxonomic studies of this species.
Supplementary Material
Supplementary Tables.docx
Revised manuscript with highlights.docx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chang HD, Ren SX. 1998. Theaceae. In: Wu, C.Y. (Ed.), Flora Reipublicae Popularis Sinicae. Science Press, p. 1–251.
- 2Chen S et al. 2023. Population genetics of Camellia granthamiana, an endangered plant species with extremely small populations in China. Front Genet. 14:1252148. 10.3389/fgene.2023.125214837867601 PMC 10585715 · doi ↗ · pubmed ↗
- 3China Ministry of Ecology and Environment (MEE). 2023. Red List of China’s Biodiversity – Higher Plants. Available online https://www.mee.gov.cn/. [accessed 2023].
- 4Choo LM, Niissalo MA, Leong PK, Khew GS. 2020. The complete plastome sequence of Gordonia penangensis Ridl. supports the transfer of Asian Gordonia into Polyspora (Theaceae). Phytotaxa. 458(2):159–166. 10.11646/phytotaxa.458.2.3 · doi ↗
- 5Dong L et al. 2021. The complete chloroplast genome of Camellia semiserrata Chi. (Theaceae), an excellent woody edible oil and landscaping species in South China. Mitochondrial DNA B Resour. 6(10):3013–3015. 10.1080/23802359.2021.197669034568567 PMC 8462872 · doi ↗ · pubmed ↗
- 6Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- 7Huang H, Shi C, Liu Y, Mao SY, Gao LZ. 2014. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 14(1):151. 10.1186/1471-2148-14-15125001059 PMC 4105164 · doi ↗ · pubmed ↗
- 8Jin JJ et al. 2020. Get Organelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. 10.1186/s 13059-020-02154-532912315 PMC 7488116 · doi ↗ · pubmed ↗