Lead Optimization: Synthesis and Biological Evaluation of Griseofulvin Derivatives as Novel SIRT6 Activators
Tilen Zorko, Jan Kogovšek, Luka Ciber, Ivana Ostojić, Nenad Maraš, Marko Novinec, Bogdan Štefane

TL;DR
Scientists developed and tested new versions of a drug called griseofulvin that can activate a protein important for aging and cell health, with some showing strong effects even at low doses.
Contribution
The study introduces novel griseofulvin derivatives with enhanced SIRT6 activation at pharmacologically relevant concentrations.
Findings
Griseofulvin itself activates SIRT6 up to 10-fold at 100 μM.
The para-1,3,4-oxadiazolephenyl analog 21 achieves 30-fold SIRT6 activation at 100 μM.
Para-tolyl derivative 24 shows potent SIRT6 activation at 10 μM.
Abstract
SIRT6, a crucial regulator of aging and cellular homeostasis, represents a promising target for small-molecule activation. In this study, we investigate griseofulvin and its derivatives as novel SIRT6 activators, focusing on the recently developed compound forvisirvat, which has progressed to Phase 2 clinical study. Biochemical evaluation revealed that griseofulvin itself possesses strong SIRT6-activating properties, achieving up to 10-fold activity at 100 μM. Modification of the griseofulvin scaffold generally led to reduced activity, prompting a focus on the oxadiazole moiety of forvisirvat. This strategy produced several analogues with higher potency, the most active at 100 μM being para-1,3,4-oxadiazolephenyl analog 21, which achieved 30-fold SIRT6 activation. Compounds bearing para-substituted phenyl rings exhibited excellent retention of activity at lower concentrations, with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1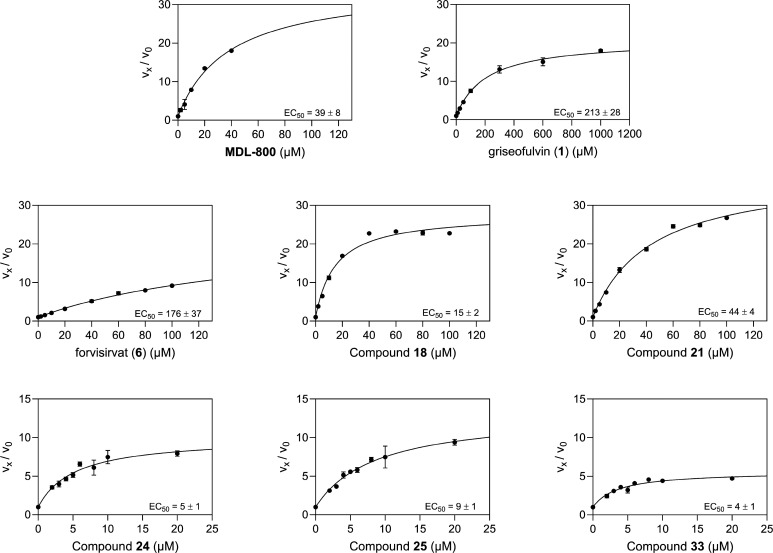 Figure 2
Figure 2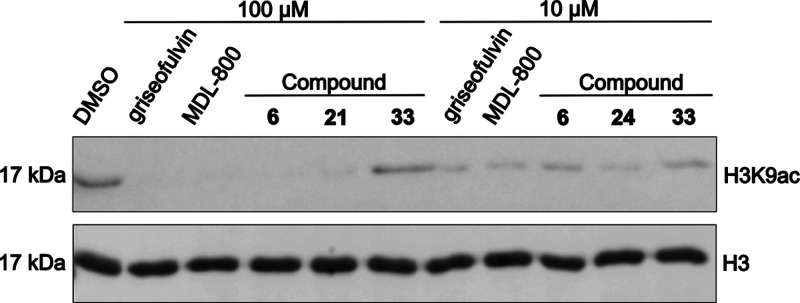 Figure 3
Figure 3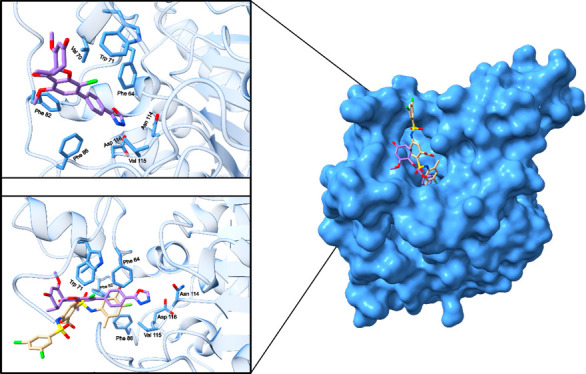 Figure 4
Figure 4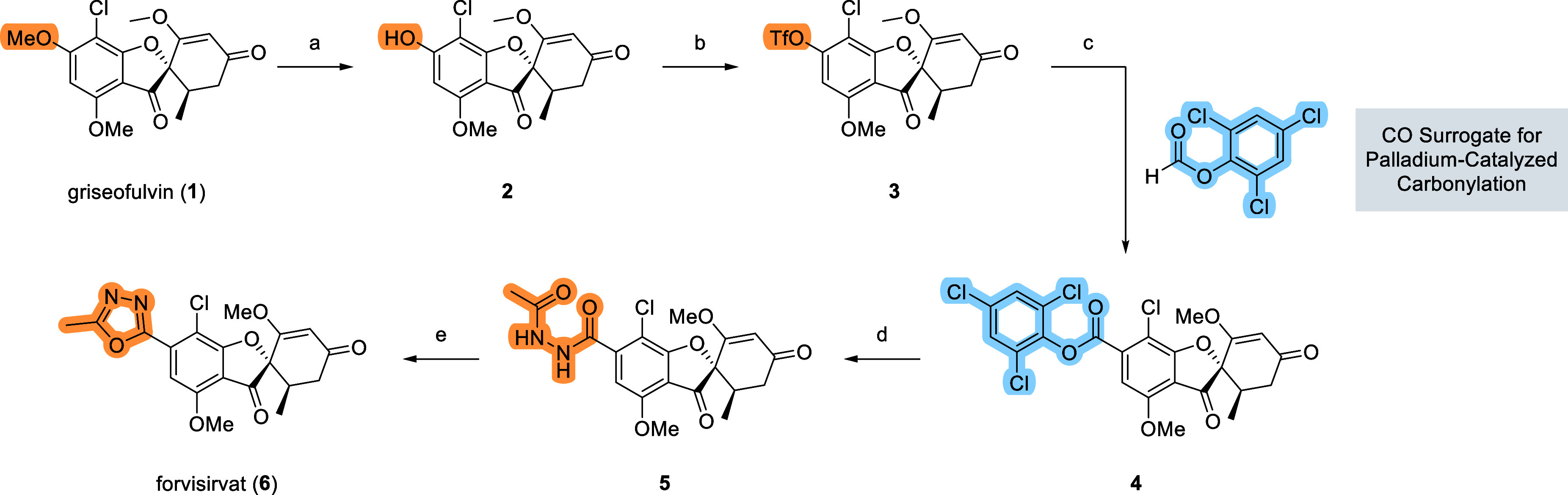 Figure 5
Figure 5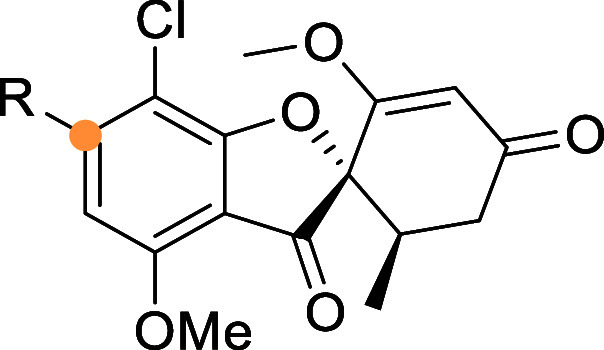 Figure 6
Figure 6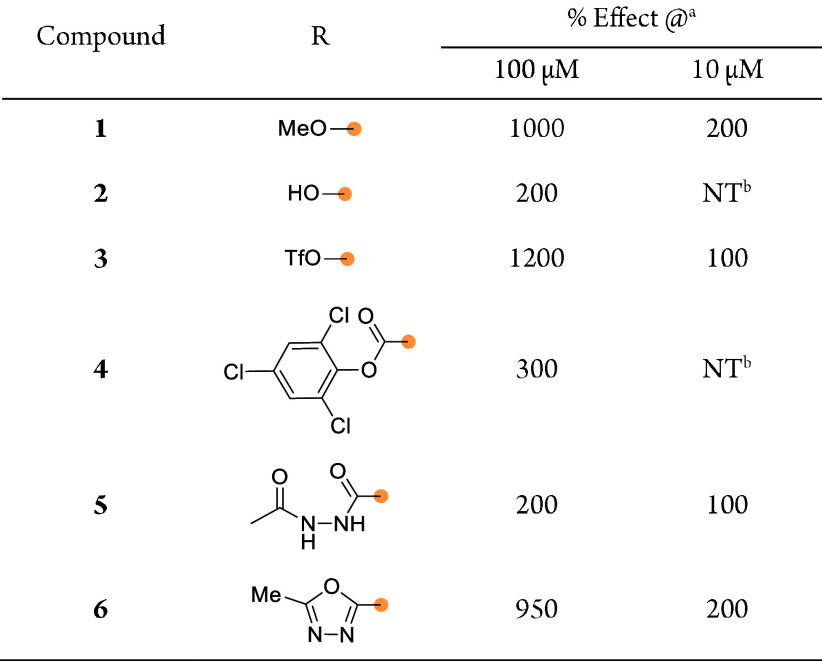 Figure 7
Figure 7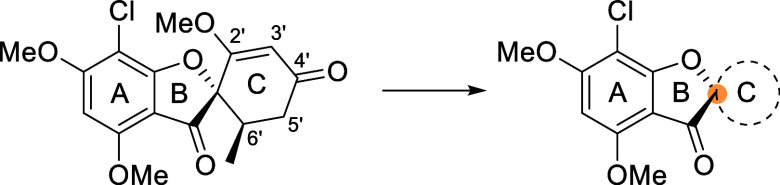 Figure 8
Figure 8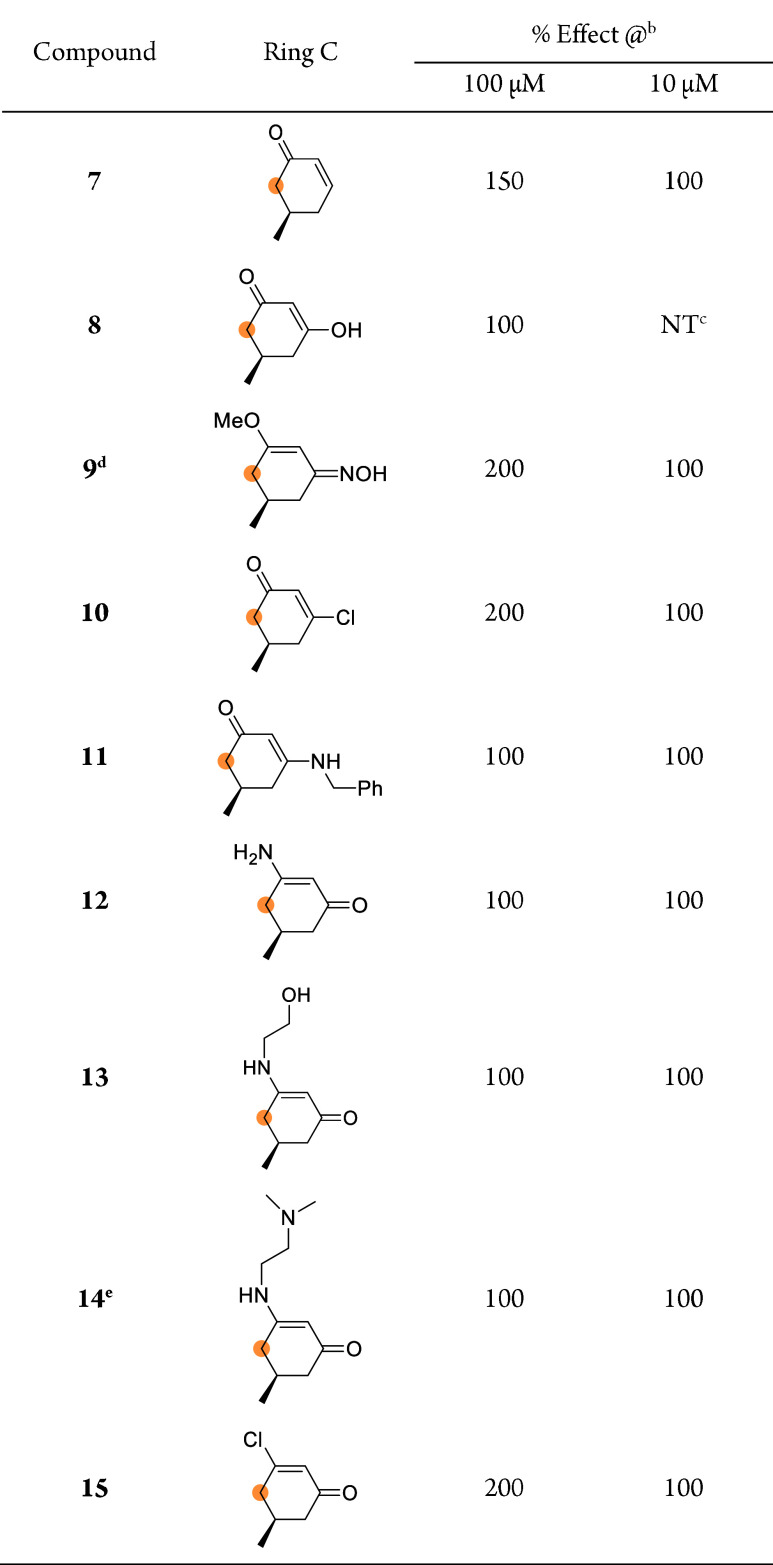 Figure 9
Figure 9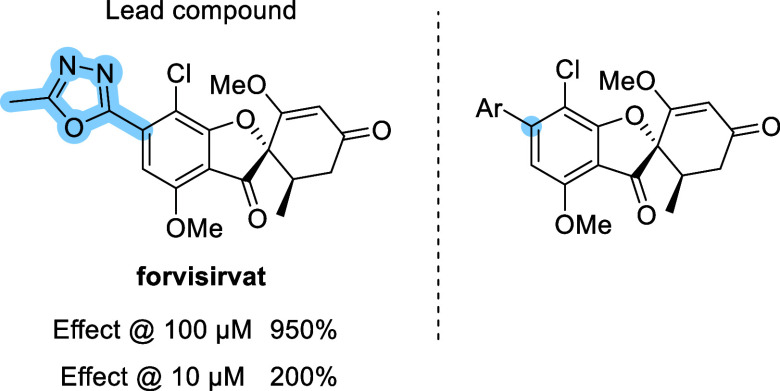 Figure 10
Figure 10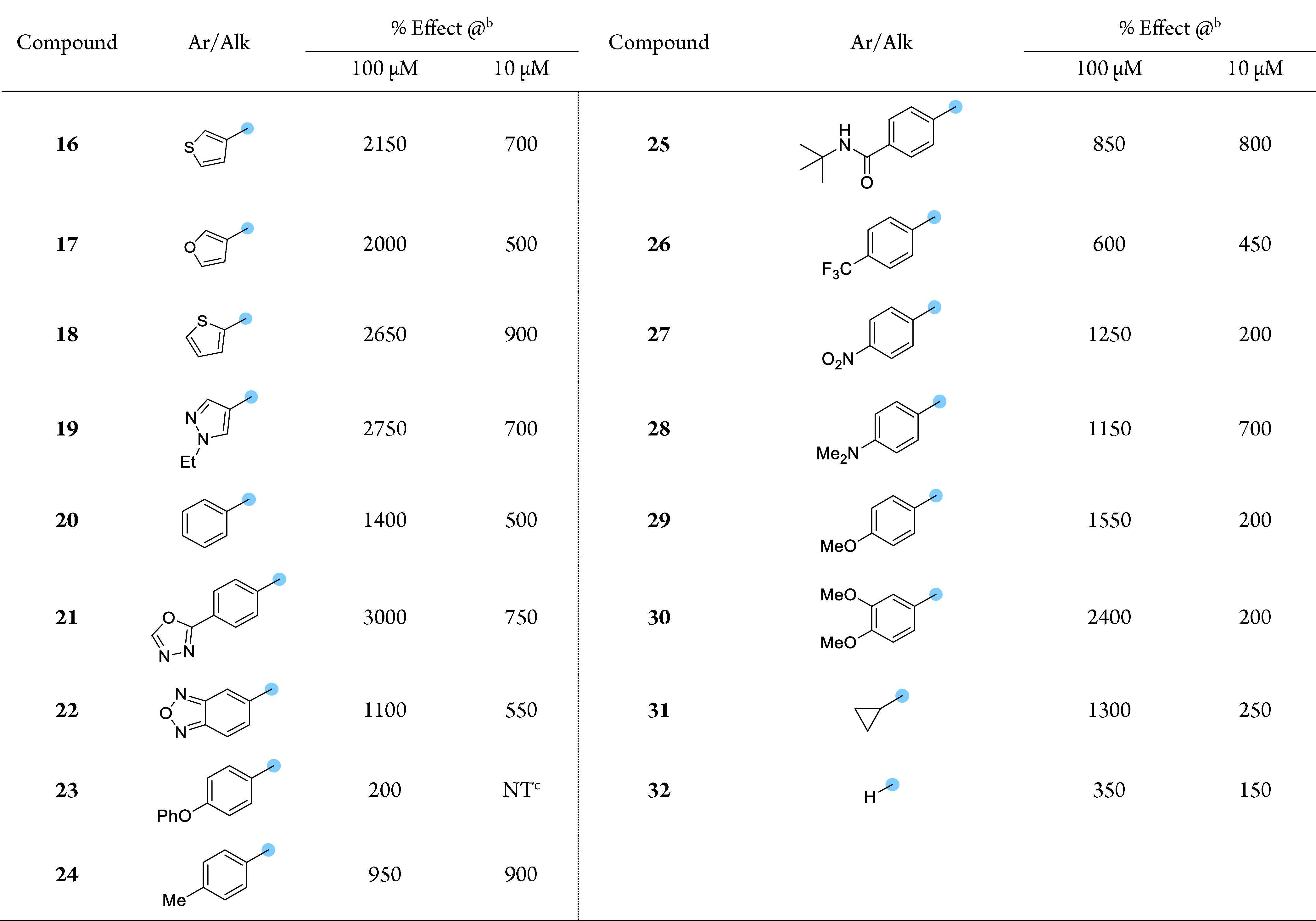 Figure 11
Figure 11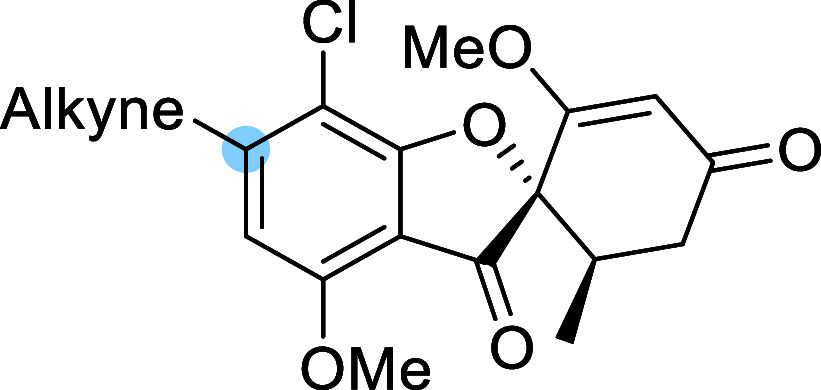 Figure 12
Figure 12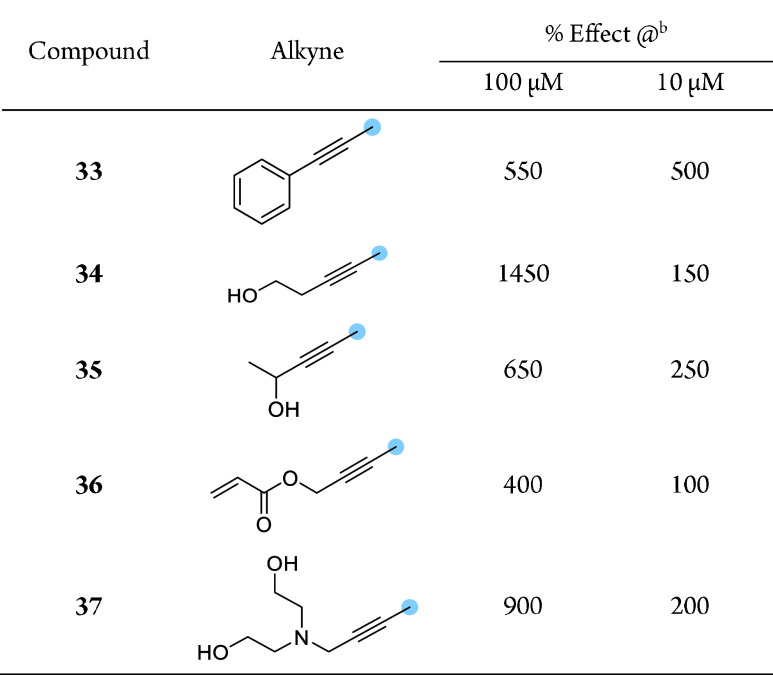 Figure 13
Figure 13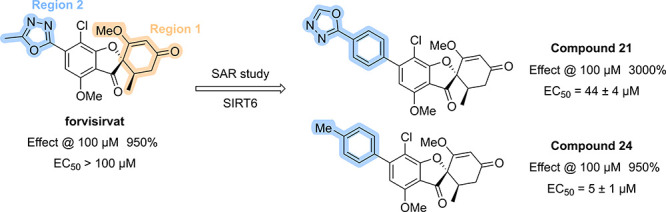 Figure 14
Figure 14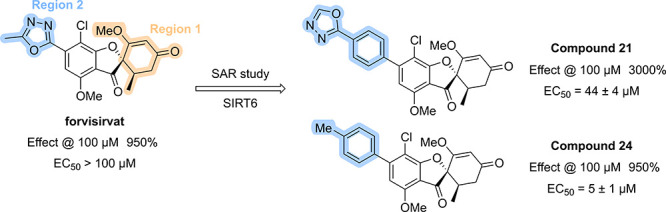 Figure 15
Figure 15- —Virella d.o.oNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSirtuins and Resveratrol in Medicine · Tea Polyphenols and Effects · Redox biology and oxidative stress
Sirtuins constitute a family of NAD^+^-dependent protein lysine deacetylases that regulate diverse physiological functions, including metabolism, the cell cycle, stress responses, and aging across both prokaryotic and eukaryotic organisms. ?,? Sirtuin regulation in mammals has been shown to delay the onset and progression of age-related diseases, such as neurodegenerative disorders, diabetes, and various forms of cancer. Mammals possess seven sirtuin isoforms (SIRT1–SIRT7), each containing a conserved NAD^+^-binding catalytic core but differing in enzymatic activity, substrate specificity, and cellular localization, resulting in distinct biological functions.? Among them, SIRT6 is a pivotal chromatin homeostasis modulator that deacetylates both histone and nonhistone proteins, including DNA repair factors and regulators of glucose metabolism.? In addition to its deacetylase activity, SIRT6 catalyzes the deacylation of long-chain fatty acid groups and exhibits mono-ADP-ribosyltransferase activity toward chromatin-silencing and DNA repair proteins, including self-mono-ADP-ribosylation.?
Functional studies in murine models have underscored the essential role of SIRT6 in maintaining metabolic balance and genomic integrity. Mice lacking SIRT6 exhibit pronounced genomic instability, disrupted glucose metabolism, and features of accelerated aging, ultimately resulting in reduced lifespan. ?,? Furthermore, SIRT6 deficiency has been linked to increased tumorigenic potential, with mutations that impair its function identified in various human cancers.? Interestingly, while SIRT6 has predominantly been characterized as a tumor suppressor, it may act as a tumor promoter in certain cellular contexts. As a tumor suppressor, SIRT6 maintains genomic integrity through its role in DNA repair and chromatin remodeling. It suppresses tumorigenesis by deacetylating histone marks such as H3K9ac and H3K56ac, thereby downregulating oncogenic transcription programs. SIRT6 also inhibits glycolysis by repressing HIF-1α activity, thus counteracting the metabolic reprogramming typical of many cancers. However, in some cancer types, elevated SIRT6 expression correlates with enhanced DNA repair capacity, allowing cancer cells to resist genotoxic stress and evade cell death. SIRT6 has also been shown to inhibit pro-apoptotic factors and support survival pathways, contributing to therapy resistance. ?,?−? ? ?
These diverse functions underscore the pivotal role of SIRT6 in aging and the maintenance of cellular homeostasis. Consequently, small molecules capable of modulating SIRT6 activity, particularly activators, are being actively investigated as promising therapeutic agents. The first synthetic activator of SIRT6 is a pyrrolo[1,2-a]quinoxaline derivative, UBCS039, which demonstrated an EC_50_ of 38 μM and increased SIRT6 activity by up to 3.5-fold in H3K9ac peptide deacetylation assays. One of the most potent SIRT6 activators identified to date is MDL-800 (Figure), a benzenesulfonamide derivative with demonstrated cellular activity. When tested on SIRT6 using the H3K9ac peptide, MDL-800 exhibited 18-fold maximal activation and an EC_50_ value of 10 μM. Its carboxylic acid analogue, MDL-801, in which the methyl carboxylate ester at position 2 of the central benzenesulfonamide ring is replaced by a carboxylic acid group, showed comparable SIRT6 activation with an EC_50_ of 6 μM. However, while MDL-800 was highly cell permeable and accumulated effectively in cells, MDL-801 exhibited poor cellular permeability and a high efflux ratio. As a result, only MDL-800 was evaluated in cellular assays. Additionally, the potency of both MDL-800 and MDL-801 remains suboptimal for therapeutic development. ?,?
Recently, forvisirvat (SP-624; Figure), a novel SIRT6 activator, advanced from Phase 1 into Phase 2 clinical trial in a study conducted by Arrivo BioVentures, making it one of the first SIRT6-targeting compounds to reach clinical evaluation. The Phase 1 study was conducted to evaluate the pharmacokinetics and safety profile of forvisirvat in healthy adults. A single Phase 2 randomized, placebo-controlled trial subsequently explored its effects in patients with major depressive disorder (MDD), with biological activity presumed to arise from activation of SIRT6. Although the trial did not meet its primary efficacy end point in the overall study population, post hoc analyses indicated a sex-dependent response, with improvement observed in female but not male participants. Forvisirvat was generally well tolerated, with no serious adverse events reported. ?−? ? To the best of our knowledge, no quantitative data on the potency of forvisirvat as a SIRT6 activator has been reported in the scientific literature. Furthermore, no structural derivatives of this scaffold have been evaluated for activity on SIRT6. As a result, its structure–activity relationship (SAR) remains unexplored. In this study, we address this gap by determining the potency of forvisirvat on SIRT6 and synthesizing a series of analogues to establish preliminary SAR insights.
To evaluate the potency of forvisirvat on SIRT6, we first synthesized the compound using a modified procedure based on a method originally developed by Daiichi Sankyo Co. (Scheme). ?,? First, griseofulvin (1) was demethylated using potassium iodide and 18-crown-6 in pyridine. The resulting phenol 2 was then triflated to afford compound 3. In the next step, formylation of the triflate was achieved using 2,4,6-trichlorophenyl formate as a highly reactive and readily available crystalline carbon monoxide surrogate in a Pd-catalyzed carbonylation reactions. The obtained trichlorophenyl ester was subsequently reacted with acetylhydrazide as a nucleophile to form product 5, which was then cyclized into forvisirvat (6) using Burgess reagent. Forvisirvat, along with all synthetic intermediates, was subsequently submitted for biological evaluation to assess activity toward SIRT6. Based on the results summarized in Table, griseofulvin itself exhibited strong SIRT6 potency, achieving 1000% activation (a 10-fold increase relative to baseline activity) at 100 μM and retaining measurable activity in the 10 μM range. Notably, demethylation of the 6-methoxy group significantly reduced activity, with the resulting phenol 2 showing only 200% activation at the same concentration. This suggests that the substitution pattern in this region of the molecule plays a critical role in SIRT6 modulation. This hypothesis was further supported by the notable increase in activity observed upon replacing the hydroxy group with a triflate, resulting in a 6-fold increase in activation of SIRT6. Such results indicate that bulky and electron-withdrawing substituents at this position are well tolerated and may even enhance activity. Additionally, the uncyclized precursor 5 in which the 1,3,4-oxadiazole ring remains in its open form, exhibited minimal activity, indicating that ring closure is essential for maintaining SIRT6 activating properties. Forvisirvat reached 950% SIRT6 activation at 100 μM, comparable to griseofulvin. At 10 μM, both compounds also displayed similar levels of activity. Building upon these initial findings, we synthesized a series of griseofulvin based analogues to further explore SAR and identify more potent SIRT6 activators.
Given the strong SIRT6-activating potency observed for griseofulvin, we sought to investigate the effect of structural modifications to its core scaffold. Several analogues of (+)-griseofulvin have been reported, typically synthesized from enantiomerically pure starting material, resulting in single stereoisomers. These derivatives can be broadly categorized into three major classes, primarily based on variations in ring C (see Table): the parent griseofulvin scaffold, griseofulvic acid (8) analogues bearing an acidic moiety at the C-ring, and isogriseofulvin analogues, which feature alternative regiochemistry within ring C. ?−? ? ? ? ? As this region contributes significantly to the molecule’s three-dimensional architecture, we explored whether structural changes to ring C would preserve or enhance activity toward SIRT6.
Reduction of griseofulvin with NaBH_4_ followed by acid-mediated elimination of water afforded product 7, which retained minimal SIRT6 activity; markedly lower than that of lead compound griseofulvin.? 4’-chlorinated analogue of isogriseofulvin 10, bearing a chlorine atom at the terminal position of the double bond, represents a structural analogue of compound 7. Despite sharing similar geometry, this analogue retained some SIRT6 activity, suggesting that additional substitution at this position may offer potential for improved activation. Replacing the chlorine with benzylamine afforded 11, which did not yield a significant improvement in activity. However, further analogues will be required to fully assess the potential of this substitution site. The 1,3-dicarbonyl substitution pattern, as present in griseofulvic acid (8), showed no meaningful activation of SIRT6. Among the synthesized analogues, compounds 9 and 12–15 maintain ring C structures closely resembling that of griseofulvin, with conserved double bond positioning and substitution patterns. In these derivatives, compound 9 features an oxime moiety replacing the carbonyl group, while compounds 12–15 bear modifications at the 2’-methoxy substituent of griseofulvin. Oxime 9 exhibited some activity toward SIRT6; however, the sample consisted of a 1:1 mixture of cis and trans isomers, and the individual isomer responsible for the activity was not determined. Regarding the substituents at the 2’ position of griseofulvin scaffold, a notable difference in activity was observed between the amine and chlorine analogues. Specifically, the chlorinated griseofulvin analogue 15 exhibited measurable SIRT6 activation, whereas its amine counterparts 12–14 were largely inactive. This underscores the critical importance of substitution at the 2’ position, with the native methoxy group in griseofulvin representing the most favorable substituent.
Since none of the ring C-modified derivatives of griseofulvin exhibited enhanced activity, we shifted our focus toward the synthesis and evaluation of forvisirvat-based analogues (Table). To access these analogues, we employed a Suzuki–Miyaura cross-coupling strategy using triflate 3 as the electrophile. Given that the lead compound features a five-membered heterocyclic ring, our initial efforts focused on synthesizing analogues bearing five-membered heteroaryl substituents. As an initial step, we tested compounds 16 and 17, featuring 3-substituted thienyl and furanyl moieties. Both compounds exhibited unexpectedly strong SIRT6 activation, significantly surpassing that of forvisirvat. To evaluate whether the position of the sulfur atom in the thiophene ring influences activity, we next tested the 2-substituted thienyl derivative 18. This compound demonstrated higher activity at both concentration levels with EC_50_ = 15 ± 2 μM (Figure), suggesting that heteroatom at the ortho-position relative to the coupling site is preferred. In contrast, the ethyl-substituted pyrazole derivative 19 showed higher activation at 100 μM, however its activity declined at lower concentration. This concentration dependent drop indicates that although some compounds achieve higher maximal activation, a steep decline in activity may limit their effectiveness at lower, more pharmacologically relevant concentrations.?
When evaluating the phenyl-substituted derivative 20, we anticipated activity comparable to that of the thiophene analogues, given the similar size and geometry of the thiophene and benzene rings. However, compound 20 exhibited lower SIRT6 activation. Next, we examined 21 containing a 1,3,4-oxadiazole moiety, a key structural feature of forvisirvat, introduced at the para-position of the phenyl ring, thus positioning the oxadiazole group further from the core griseofulvin scaffold. Compound 21 demonstrated the highest SIRT6 activation observed in the entire series, reaching 30-fold activation at 100 μM. However, its activity declined at lower concentrations, corresponding to an EC_50_ value of 44 ± 4 μM. When the oxadiazole moiety was fused to a benzene ring in the form of a 2,1,3-benzoxadiazole system, the resulting compound 22 exhibited reduced activity, with SIRT6 activation dropping to 1100% at 100 μM. The overall performance of the para-substituted compounds, particularly compound 21, underscored their potential and motivated further exploration of structural modifications at this position. Replacement of the oxadiazole moiety with a phenoxy group (compound 23) resulted in a substantial reduction in activity, suggesting that large, freely rotating substituents at this position may be sterically unfavorable.
Among the para-substituted analogues evaluated, compounds 24 and 25 stood out due to their exceptional retention of potency at lower concentrations. Although their effects at 100 μM were modest compared to other analogues, only a slight decrease of activity was observed at 10 μM. This observation is further supported by their measured EC_50_ values of 5 ± 1 μM and 9 ± 1 μM (Figure) respectively, indicating that both compounds may exhibit efficacy at pharmacologically relevant concentrations. Encouraged by these results, we synthesized a series of para-substituted analogues incorporating both electron-donating and electron-withdrawing groups. However, all synthesized derivatives ultimately lost most of their potency at 10 μM concentration. At 100 μM, the trifluoromethyl-substituted analogue 26 exhibited lower activity than compound 24. While the trifluoromethyl group is slightly bulkier and more electron-withdrawing than a methyl group, its size alone is unlikely to account for the observed loss in activity, as bulkier substituents, such as the 4-(tert-butylcarbamoyl) group in analogue 25, are well tolerated within the binding pocket.? Compound 30 features substitutions at both the para and meta position, demonstrated improved activity over the mono-para-substituted methoxyphenyl analogue 29 at 100 μM. However, this advantage was not retained at lower concentrations, as both compounds showed a loss of activity at 10 μM. Overall, very few analogues maintained high potency across both concentration ranges, highlighting the rarity of this characteristic and its importance in the identification of promising drug candidates.
To access alkyl-substituted variants of forvisirvat, we employed a Suzuki–Miyaura coupling with cyclopropyl boronic acid, yielding compound 31, which surprisingly, exhibited comparable potency profiles to forvisirvat. This suggests considerable substituent tolerance at this position. However, as demonstrated by the low activity of the unsubstituted product 32 and the previously reported 6-O-demethyl griseofulvin (2), the presence of an appropriate substituent at the 6-position of the griseofulvin scaffold appears to be essential for SIRT6 activation.
To further explore the substituent tolerance at the 6-position of the griseofulvin scaffold, we next introduced alkynyl groups using a Sonogashira cross-coupling strategy (Table). The benchmark compound 33, bearing a phenylacetylene moiety, exhibited moderate SIRT6 activation at 100 μM but retained much of its activity at 10 μM, affording the lowest EC_50_ among all synthesized compounds (4 ± 1 μM). It was the only member of the alkyne series to maintain such high potency at the lower concentration, mirroring trends observed in the heteroaryl substituted series, where certain phenyl-containing analogues also demonstrated strong retention of activity. Next, a series of alkynes featuring a polar group at the terminal position were tested. Among these, compound 34 showed the highest activation in the series, reaching approximately 14-fold SIRT6 activation at 100 μM, whereas the branched isomer 35, with an additional methyl group at the β-position, displayed lower overall potency. Compound 37, which features a bulky, hydroxylated polar side chain, retained substantial activity at both concentrations, further indicating that the SIRT6 binding pocket can accommodate large, polar substituents.
To evaluate whether the synthesized compounds could deacetylate a chromatin substrate, which is the endogenous target of SIRT6, we performed an enzyme assay using isolated nucleosomal histones instead of a synthetic peptide (Figure). A control experiment was conducted using DMSO alone. MDL-800 was included as a literature standard, as it is known to promote deacetylation of native histones.? All tested compounds displayed histone deacetylation activity at 100 μM, including the lead compounds griseofulvin and forvisirvat, confirming their ability to act on native chromatin substrates. Among them, 33 produced the weakest deacetylation response relative to the DMSO control. At 10 μM, deacetylation activity was still observed for all compounds; however, forvisirvat (6) displayed lower deacetylation activity than compound 24, which, under these conditions, resulted in near-complete deacetylation of histones. This observation correlates well with their relative activities at 10 μM, where forvisirvat showed only 200% activation, whereas compound 24 was the most active analogue, reaching 900% activation at this concentration. Interestingly, compound 33, which exhibited the lowest half maximal effective concentration among the synthesized analogues (EC_50_ = 4 ± 1 μM) and showed 500% activity at 10 μM, also displayed weaker histone deacetylation activity than compound 24.
Molecular docking was used to predict the binding mode of compound 21 with SIRT6, as shown in Figure. Compound 21 occupies the allosteric pocket, which is very similar to the binding sites reported for UBCS039? and MDL-801? (see Figure, showing a superposition of 21 and MDL-801). The aliphatic moiety of compound 21 lies at the entrance of the channel, in close proximity to Val70 and Trp71. The phenyl ring of the 4-(oxadiazolyl)phenyl substituent is spatially aligned with the 5-bromo-4-fluoro-2-methylaniline moiety of MDL-801 in the superimposed structure and therefore engages in interactions with the same amino acid residues, namely Phe64, Val70, Phe82, Phe86, and Val115. However, the oxadiazole ring of compound 21 extends further into the binding pocket than MDL-801 (see Figure), placing one of the oxadiazole nitrogen atoms in close proximity (≈ 3.8 Å) to Asn114 and Asp116. Interactions involving these two amino acid residues in SIRT6 have been described in the literature; however, they have been reported in the context of inhibition rather than activation, notably for the inhibitor trichostatin A.?
In conclusion, this study describes the lead optimization of griseofulvin based scaffolds as small molecule activators of SIRT6. Structure activity relationship analysis identified key structural features required for activity and yielded analogues that retained substantial SIRT6 activation at micromolar concentrations. Selected compounds were further validated using a native histone substrate, supporting their functional activity beyond peptide-based assays. Although isoform selectivity and cellular activity remain to be established, these findings provide a foundation for further optimization of SIRT6-targeted activators and support continued exploration of this chemotype.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Houtkooper R. H.Pirinen E.Auwerx J.Sirtuins as Regulators of Metabolism and Healthspan Nat. Rev. Mol. Cell Biol.201213422523810.1038/nrm 329322395773 PMC 4872805 · doi ↗ · pubmed ↗
- 2Imai S.Armstrong C. M.Kaeberlein M.Guarente L.Transcriptional Silencing and Longevity Protein Sir 2 Is an NAD-Dependent Histone Deacetylase Nature 2000403677179580010.1038/3500162210693811 · doi ↗ · pubmed ↗
- 3Chalkiadaki A.Guarente L.The Multifaceted Functions of Sirtuins in Cancer Nat. Rev. Cancer 2015151060862410.1038/nrc 398526383140 · doi ↗ · pubmed ↗
- 4Klein M. A.Denu J. M.Biological and Catalytic Functions of Sirtuin 6 as Targets for Small-Molecule Modulators J. Biol. Chem.202029532110211104110.1074/jbc.REV 120.01143832518153 PMC 7415977 · doi ↗ · pubmed ↗
- 5Liszt G.Ford E.Kurtev M.Guarente L.Mouse Sir 2 Homolog SIRT 6 Is a Nuclear ADP-Ribosyltransferase J. Biol. Chem.200528022213132132010.1074/jbc.M 41329620015795229 · doi ↗ · pubmed ↗
- 6Kanfi Y.Naiman S.Amir G.Peshti V.Zinman G.Nahum L.Bar-Joseph Z.Cohen H. Y.The Sirtuin SIRT 6 Regulates Lifespan in Male Mice Nature 2012483738821822110.1038/nature 1081522367546 · doi ↗ · pubmed ↗
- 7Mostoslavsky R.Chua K. F.Lombard D. B.Pang W. W.Fischer M. R.Gellon L.Liu P.Mostoslavsky G.Franco S.Murphy M. M.Mills K. D.Patel P.Hsu J. T.Hong A. L.Ford E.Cheng H.-L.Kennedy C.Nunez N.Bronson R.Frendewey D.Auerbach W.Valenzuela D.Karow M.Hottiger M. O.Hursting S.Barrett J. C.Guarente L.Mulligan R.Demple B.Yancopoulos G. D.Alt F. W.Genomic Instability and Aging-like Phenotype in the Absence of Mammalian SIRT 6Cell 2006124231532910.1016/j.cell.2005.11.04416439206 · doi ↗ · pubmed ↗
- 8Kugel S.Feldman J. L.Klein M. A.Silberman D. M.Sebastián C.Mermel C.Dobersch S.Clark A. R.Getz G.Denu J. M.Mostoslavsky R.Identification of and Molecular Basis for SIRT 6 Loss-of-Function Point Mutations in Cancer Cell Rep.201513347948810.1016/j.celrep.2015.09.02226456828 PMC 4618237 · doi ↗ · pubmed ↗