Targeting Protein Tyrosine Phosphatase 1B (PTP1B) to Improve Insulin Sensitivity Using Indole-Fused Glycyrrhetinic Acid Conjugates with Amino Acids
Ledy De-la-Cruz-Martínez, David Equihua-González, Diana Laura Torres-Chacón, Erandi Ortiz-Barragán, J. Martin Torres-Valencia, Rubria Marlen Martínez-Casares, Jaime Pérez-Villanueva, Martín González-Andrade, Julio César Almanza-Pérez, Francisco Cortés-Benítez

TL;DR
This study improves insulin sensitivity by developing new PTP1B inhibitors that show strong activity and potential for treating type 2 diabetes.
Contribution
New indole-fused glycyrrhetinic acid conjugates with enhanced PTP1B inhibition and improved insulin sensitivity in mice.
Findings
Compounds 5a and 5d showed up to 4-fold greater PTP1B inhibition with submicromolar IC50 values.
Compounds 5a and 5b increased GLUT4 receptor mRNA expression in C2C12 myoblast cells.
The compounds reduced glucose levels in streptozotocin-induced diabetic mice during insulin tolerance tests.
Abstract
Protein Tyrosine Phosphatase 1B (PTP1B) is a crucial enzyme that significantly modulates insulin and leptin signaling, making it a highly promising target for the treatment of type 2 diabetes (T2D). We previously reported the synthesis and inhibitory activity of FC-114, an indole-fused glycyrrhetinic acid derivative that potently inhibits PTP1B. In this study, we synthesized four FC-114 conjugates with amino acids at the C30 position to enhance their inhibitory activity against PTP1B in vitro. The results indicated that incorporating glycine (compound 5a) and serine (compound 5d) substantially enhanced inhibitory activity against PTP1B, achieving up to 4-fold greater potency, with submicromolar IC50 values of 0.64 and 0.54 μM, respectively. Inhibitory assessments of the short form (hPTP1B1‑285) and long form (hPTP1B1‑400) of PTP1B, along with enzymatic kinetics studies, molecular…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
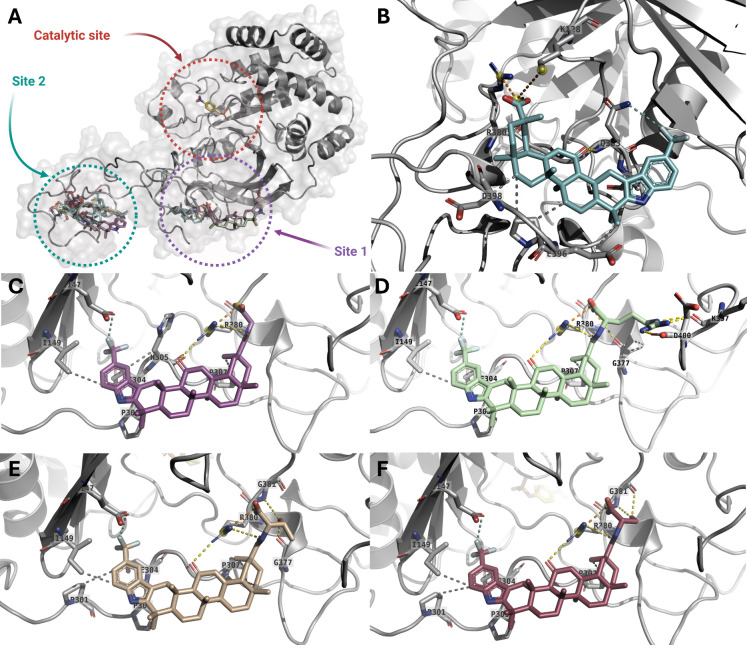 Figure 1
Figure 1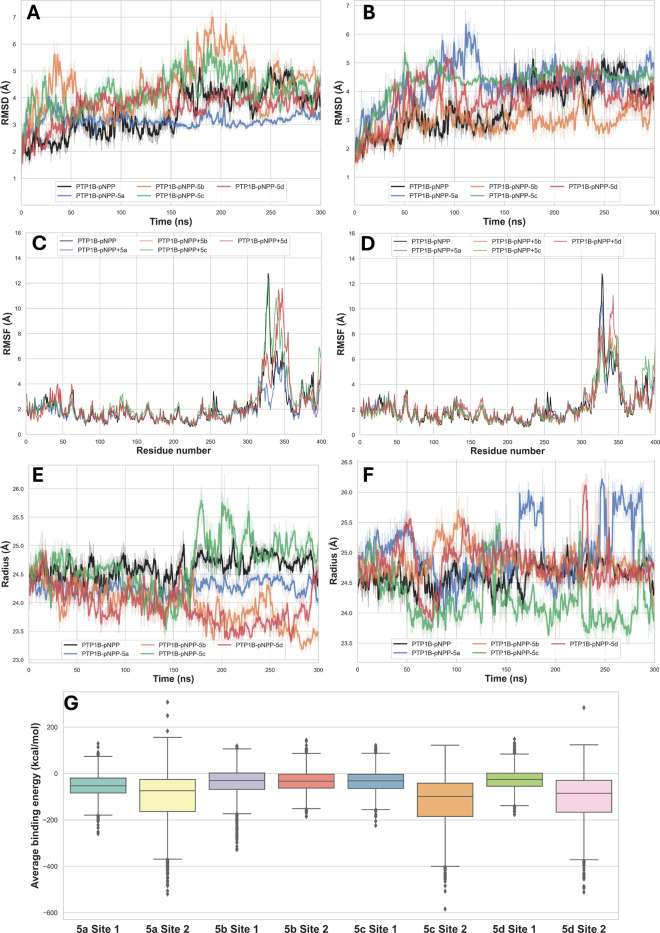 Figure 2
Figure 2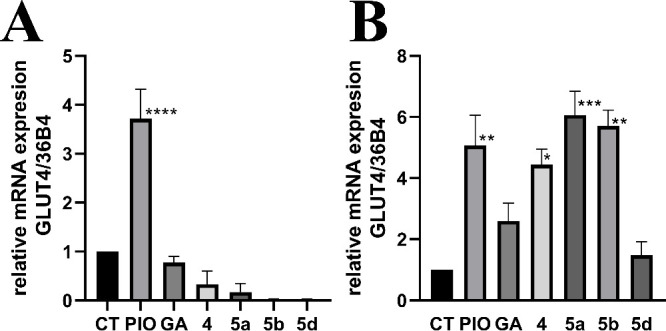 Figure 3
Figure 3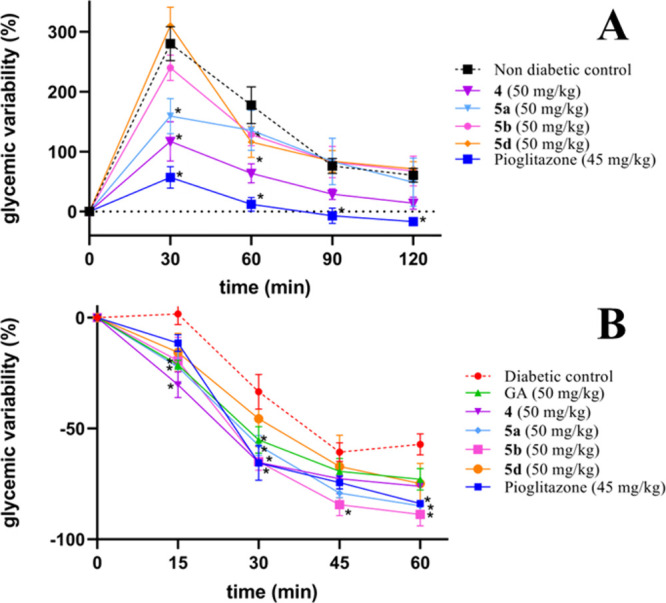 Figure 4
Figure 4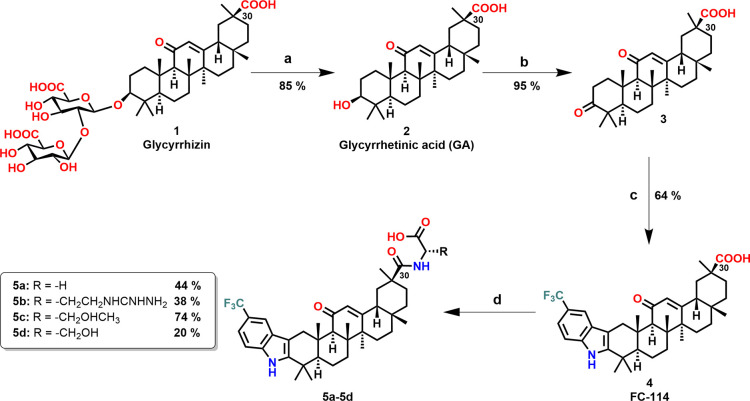 Figure 5
Figure 5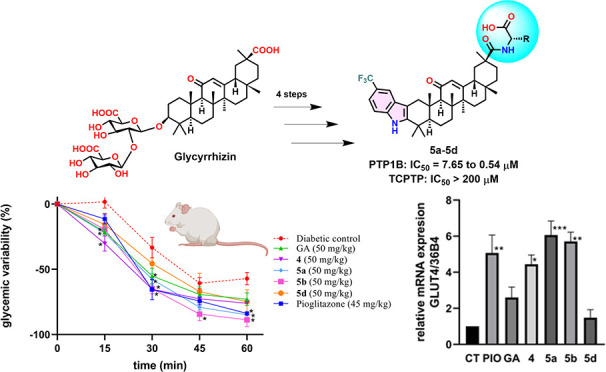 Figure 6
Figure 6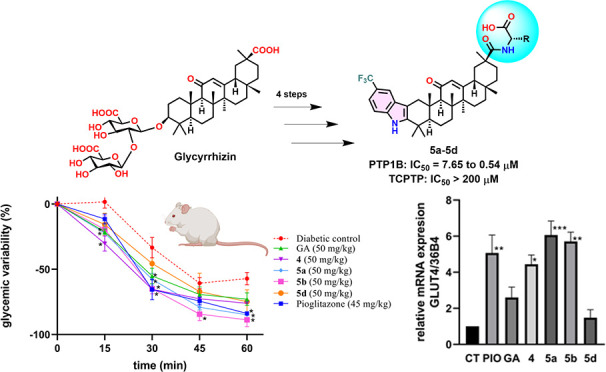 Figure 7
Figure 7- —Consejo Nacional de Humanidades, Ciencias y Tecnolog?as10.13039/501100003141
- —Direcci?n General de Asuntos del Personal Acad?mico, Universidad Nacional Aut?noma de M?xico10.13039/501100006087
- —Direcci?n General de Asuntos del Personal Acad?mico, Universidad Nacional Aut?noma de M?xico10.13039/501100006087
- —DGTIC-UNAMNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtein Tyrosine Phosphatases · Glycogen Storage Diseases and Myoclonus · Diabetes and associated disorders
Diabetes represents one of the fastest-growing global health challenges, as evidenced by 2024 estimates showing 635 million individuals with impaired glucose tolerance and 488 million with impaired fasting glucose, along with over 3.4 million adults (aged 20–79) who died from related complications.? The disorder is a chronic metabolic condition characterized by elevated blood sugar levels due to insufficient insulin production, insulin deficiency, or decreased insulin sensitivity. Type 2 diabetes (T2D), which accounts for over 90% of all global cases, is the primary concern and is projected to become the second leading cause of global disease burden by 2050. ?,? Its characteristic pathology begins with insulin resistancethe reduced responsiveness of peripheral tissues to insulinwhich drives compensatory hyperinsulinemia. This excessive demand eventually stresses the system, progressing to irreversible pancreatic β-cell dysfunction and subsequent impaired insulin secretion. ?,? The treatments for T2D include sulphonylureas, α-glucosidase inhibitors, thiazolidinediones, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 receptor (GLP-1R) and gastric inhibitory peptide (GIP) agonists, as well as sodium-glucose cotransporter 2 (SGLT2) inhibitors. However, adverse effects, suboptimal efficacy, or lack of selectivity limit their use. ?,?
In this context, Protein Tyrosine Phosphatase 1B (PTP1B) is a well-recognized target of T2D, as its inhibition enhances insulin sensitivity and improves glucose homeostasis. Unfortunately, many PTP1B inhibitors lack selectivity and also inhibit closely related phosphatases, such as T-cell Protein Tyrosine Phosphatase (TCPTP), leading to undesirable side effects. ?−? ? ? ? ? Therefore, to date, no PTP1B inhibitors have progressed beyond Phase II clinical trials. ?,? We previously reported compound FC-114 (4), an indole-fused derivative of glycyrrhetinic acid, which demonstrated potent PTP1B inhibition in vitro and improved glucose metabolism in streptozotocin (STZ)-induced diabetic rats. ?,?,? However, the in vivo effect was less significant than expected, likely due to the compound’s high lipophilicity, which may have limited its aqueous solubility and overall bioavailability. To mitigate these limitations, we synthesized derivatives of compound 4 that are conjugated to amino acids at position C30, including Gly (5a), Arg (5b), Thr (5c), and Ser (5d). The strategy of amino acid conjugation is frequently employed in medicinal chemistry due to its numerous advantages: it enhances structural diversity to improve interactions with targets, increases aqueous solubility, and reduces toxicity. ?,? The compounds 5a, 5b, 5c, and 5d were synthesized following the procedure outlined in Scheme. Glycyrrhizin was used as a raw material and hydrolyzed to produce Glycyrrhetinic acid (GA, 2). The secondary alcohol at the C3 position of GA was then oxidized to yield 3-oxoglycyrrhetinic acid (3). Subsequently, the Fischer indole synthesis was employed to react this intermediate with 4-(trifluoromethyl)phenylhydrazine, yielding compound 4. For the synthesis of amino acid conjugates, compound 4 was reacted with the methyl esters of l-amino acids Gly, Arg, Thr, and Ser using 1,1'-carbonyldiimidazole (CDI) as a coupling agent, followed by alkaline hydrolysis, affording compounds 5a–5d in moderate yields.
The structures of the final compounds were confirmed by ^1^H and ^13^C NMR, along with COSY, HMBC, and HSQC experiments (Supporting Information Figures S1–S20). The chemical shift data of the indole ring as well as the triterpenoid skeleton of these compounds are in accordance with the data of compound 4. ?,?,? However, in the ^1^H NMR spectra, we found some representative signals in the aromatic region from 7.22 to 7.91 ppm characteristic of NH signal of amide group (5a (7.91 ppm, t, J = 5.8 Hz), 5b (7.70, d, J = 7.6 Hz), 5c (6.99 ppm, d, J = 6.1 Hz) and 5d (7.22, d, J = 6.1 Hz)). On the other hand, doublets of doublets signals with J = 17.8, 5.3, and 17.3, 5.5 Hz at 3.80 and 3.67 ppm, respectively, were assigned to Hα from glycine in the 5a compound. For the 5b compound, there are two broad singlets at 7.81 and 7.00 ppm assigned to NH from the guanidine group. Additionally, a multiplet at 4.23 ppm is assigned to Cα, and a signal at 156.88 ppm was assigned to Cε of arginine. Furthermore, multiple and singlet signals at 3.95 (1H) and 1.78 (3H) ppm, respectively, corresponding to Cα and Cγ from threonine in the compound 5c. Finally, a broad singlet (4.56 ppm) and a multiple signal (4.06–3.96 ppm) were assigned to OH and Cα from serine in derivative 5d.
The compounds 5a–5d, and positive controls GA, Ursolic acid (UA), Ertiprotafib, and Sodium Orthovanadate (SOV) were assessed for their inhibitory effect on the long form of PTP1B enzyme (hPTP1B_1‑400_), to prevent the formation of p-nitrophenol from p-nitrophenylphosphate (pNPP). Several concentrations of new derivatives and controls were assessed, giving IC_50_ values ranging from 7.65 to 0.54 μM (Table). The potency order of the indole derivatives of GA is as follows: 5d > 5a > 5c > 5b. Notably, compounds 5a (IC_50_ = 0.64 μM) and 5d (IC_50_ = 0.54 μM) are 4-fold more potent than compound 4 (IC_50_ = 2.28 μM) and are also significantly more potent than GA (IC_50_ = 56.49 μM), being 105-fold and 88-fold more potent, respectively. Additionally, both compounds are 12-fold and 10-fold more potent than UA, a pentacyclic triterpene widely reported as a PTP1B inhibitor. Moreover, they demonstrated 3-fold greater potency than the gold-standard PTP1B inhibitors Ertiprotafib (IC_50_ = 1.73 μM) and SOV (IC_50_ = 1.87 μM). Ertiprotafib is a PTP1B inhibitor that reached phase II clinical trials for the treatment of T2D ?,? while SOV is a potent, nonselective, and commonly used competitive inhibitor of PTP1B.? In contrast, compounds 5b (IC_50_ = 7.65 μM) and 5c (IC_50_ = 3.42 μM) exhibited 3.3-fold and 1.5-fold lower potency than 4, respectively.
Several interesting observations emerged from the results regarding structure–activity relationships. For example, the addition of an amino acid at position C30 affects the inhibitory activity against PTP1B. Specifically, when glycine or serine is added, inhibitory activity increases, whereas the addition of threonine or arginine decreases activity. This indicates that smaller amino acid side chains are more favorable for enhancing inhibitory activity. Furthermore, arginine contains an ionizable guanidino group at physiological pH, which may contribute to its reduced inhibitory activity against PTP1B. In contrast, serine and threonine are polar amino acids containing primary and secondary alcohols, respectively. However, threonine also carries a methyl group that introduces steric hindrance, which likely contributes to its reduced inhibitory activity against PTP1B compared to serine.?
To indirectly explore the role of the disordered C-terminal region of PTP1B in the binding of synthesized compounds, we evaluated compounds 5a, 5b, and 5d against the short form of PTP1B (hPTP1B_1–285_), which lacks this region (Supporting Information, Figure S29). This intrinsically disordered C-terminal domain of PTP1B (residues 301–400) is specific to PTP1B.? In contrast, the catalytic domain of PTP1B shares a high degree of homology with other protein tyrosine phosphatases (PTPs), such as TCPTP. The results indicated that compounds 4, 5a, and 5d displayed a 6- to 5-fold greater potency against hPTP1B_1‑400_ compared to against hPTP1B_1‑285_. This suggests that the disordered C-terminus of PTP1B is essential for the inhibitory activity, indicating that the primary binding region for these compounds is likely located at this site. This observation aligns with previous research, including studies on Trodusquemine (MSI-1436), an aminosterol known for its ability to inhibit PTP1B by binding to the C-terminal site.? In contrast, compound 5b was found to be 3.5-fold more potent against hPTP1B_1‑285_ than against hPTP1B_1‑400_. The presence of Arg at the C30 position of FC-114 suggests a reduced affinity of the compound for the longer form of PTP1B. The guanidine group of Arg carries a positive charge at physiological pH and can interact with negatively charged regions, particularly those around Glu or Asp residues, within the catalytic domain of PTP1B.
The catalytic domain of PTP1B shares 74% sequence homology with that of TCPTP, which plays a crucial role in immune function.? To evaluate the selectivity of compounds 5a–5d, we tested them against hTCPTP_1‑415_. As shown in Figure S30 of the Supporting Information, the results indicated that none of the synthesized compounds displayed any inhibitory activity against TCPTP, even at concentrations of up to 200 μM. Consequently, these compounds are at least 26- to 370-fold selective for PTP1B over TCPTP.
To assess their mode of inhibition on hPTP1B_1–400_, compounds 5a, 5b, and 5d were selected for enzyme kinetic assays (Table). The Lineweaver–Burk plots are illustrated in Figure S31 of the Supporting Information. The PTP1B inhibitors 5a, 5b, and 5d exhibited K i values of 1.15, 0.79, and 1.91 μM, respectively. Furthermore, these compounds demonstrated uncompetitive inhibition, suggesting that they may bind to an allosteric site on PTP1B.
Based on the inhibition types observed for compounds 5a–5d, we explored their potential binding modes using a three-dimensional model of the PTP1B_1‑400_ enzyme complexed with the pNPP substrate (PTP1B_1‑400_-pNPP) previously reported by our group. ?,? Given that the C-terminal region of PTP1B is intrinsically disordered and that the full-length structure is based on an AlphaFold prediction, the following docking analyses are intended to provide qualitative, hypothesis-generating insights rather than definitive structural conclusions. Molecular docking simulations were conducted using three software programs: AutoDock 4.2 (The Scripps Research Institute, La Jolla, CA, USA), Vina (The Scripps Research Institute, La Jolla, CA, USA), and GOLD (The Cambridge Crystallographic Data Centre, Cambridge, UK).
To identify the preferred binding sites for the synthesized compounds within the PTP1B_1–400_-pNPP complex, a blind docking simulation was conducted using AutoDock. The resulting docking poses suggested that the compounds may preferentially sample two main regions within the enzyme, with a higher frequency of poses located in the disordered C-terminal region rather than in the catalytic domain (FigureA). The first site (Site 1) is a pocket that includes a portion of the catalytic domain and a significant portion of the unstructured C-terminal region, featuring the residues Lys^128^, Ile^134^, Glu^147^, Ile^149^, Pro^303^, Glu^304^, Ile^306^, Pro^307^, Arg^380^, Ala^382^, Gln^383^, Ala^384^, Pro^387^, Pro^395^, Lys^397^, and Asp^400^. The second site (Site 2) is situated in the unstructured C-terminal region, consisting of the residues: His^320^, Glu^337^, Asp^341^, Cys^344^, Pro^345^, Lys^350^, Ser^352^, Pro^353^, Ala^356^, Pro^358^, and Arg^371^.
Subsequently, site-directed docking simulations of the GA derivatives were performed at Sites 1 and 2 using AutoDock, Vina, and GOLD (Supporting Information Table S1). The docking analyses indicated that the newly synthesized compounds generally achieved more favorable docking scores at Site 1 (particularly with AutoDock and ChemPLP) compared to compound 4. Across all three docking programs, the top-ranked poses of compounds 5a–5d at Site 1 adopted broadly similar orientations, with the main differences arising from the orientation of the amino acid side chains attached at the C30 position (FigureC-?F and Supporting Information, Figures S32A–S32E). In contrast, compound 4 adopted a distinct docking pose within this region (FigureB).
Inspection of the top-ranked docking poses suggested a network of potential hydrophobic, hydrogen-bonding, and ionic interactions that may contribute to the observed binding trends. For compounds 5a–5d, the triterpenoid scaffold and indole moiety were positioned in proximity to Pro^303^, Pro^307^, and Ile^149^, consistent with hydrophobic contacts. In addition, the trifluoromethyl group was oriented toward Glu^147^ and Glu^304^, allowing for potential halogen interactions, while the carbonyl groups at C11 and C30 were positioned to act as hydrogen bond acceptors for Arg^380^. In contrast, compound 4 was oriented such that its triterpenoid scaffold performed hydrophobic interactions with Ile^134^, Ala^383^, Pro^384^, Pro^387^, and Pro^395^, while its trifluoromethyl and C11 carbonyl groups were positioned near Gln^393^ to form H-bonds. The incorporation of amino acids at C30 in compounds 5a–5d increased the number of potential hydrogen-bonding or ionic interactions; for example, the amide group in compound 5a form an H-bond with Gly^377^, the hydroxyl groups in compounds 5c and 5d acted as H-bond donors with Gly^377^ and Gly^381^, respectively, and the guanidinium group in compound 5b was oriented toward Asp^400^ and Lys^397^ forming salt bridge interactions.
At Site 2, the docking analyses suggested that the amino acid-conjugated derivatives generally achieved more favorable docking scores with AutoDock and GOLD (ChemPLP), with the exception of compound 5c, compared to compound 4. The top-ranked poses of compounds 5a–5d adopted orientations opposite to that of compound 4 within this region (Supporting Information, Figures S32F–S32J). As observed for Site 1, amino acid conjugation increased the number of potential hydrogen-bonding and ionic interactions. For instance, the carboxylate group of compound 4 was positioned near His^320^ and Asn^321^, whereas in compound 5a, the newly introduced amide group forms H-bonds with Asn^321^ and Pro^358^. In compounds 5c and 5d, the hydroxyl groups were positioned to form H-bonds with Glu^336^, Glu^337^, and Asp^341^, as well as Gln^332^, Val^334^, and Glu^336^, respectively, while the guanidinium group of compound 5b was oriented toward Glu^336^ and Asp^341^ to form salt-bridge interactions as well as an H-Bond with Ser^352^.
To gain a better understanding of the stability of the PTP1B_1–400_-pNPP-ligand complexes resulting from molecular docking simulations, we conducted molecular dynamics simulations using YASARA Structure ?,? program and AMBER 11 force field (FiguresA–?G and Supporting Information Table S2). Overall, the trajectory of 300 ns simulations reveals a clear site-dependent modulation of protein dynamics and ligand accommodation, highlighting differences that are not fully captured by binding free-energy estimates. At Site 2, ligand binding induced variable degrees of global conformational deviation. While 5b displayed the lowest average Cα RMSD, suggesting limited structural perturbation, this ligand was associated with comparatively weak binding energetics. In contrast, 5a, 5c, and 5d induced larger conformational adjustments, reflected by higher RMSD values, consistent with a binding mode that requires protein reorganization. Among these ligands, 5c exhibited the most favorable MM/PBSA binding free energy; however, this energetic advantage was accompanied by increased structural deviation and did not translate into a clear stabilization of protein dynamics, as indicated by RMSF analysis. These observations suggest that strong binding energetics at Site 2 may arise from transient or highly adaptive interactions rather than from a consistently stable binding mode.
In contrast, Site 1 displayed a distinct dynamic profile characterized by improved structural stabilization upon ligand binding. Notably, complexes formed with 5a and 5d displayed radius of gyration values comparable to that of the PTP1B_1–400_-pNPP protein, indicating preservation of the overall protein compactness upon ligand binding and suggesting minimal disruption of the global fold. Moreover, 5a showed the lowest RMSD and the lowest RMSF among the evaluated ligands at this site, indicating a stabilizing effect on both global and local protein motions. This behavior was accompanied by the most favorable binding free energy at Site 1, suggesting a binding mode that is energetically favorable while minimizing conformational strain on the protein. 5d, although associated with less favorable MM/PBSA energies, exhibited a dynamic behavior comparable to that of 5a, maintaining moderate RMSD values and avoiding excessive local flexibility. This combination suggests a binding mode that is dynamically well tolerated by the protein, potentially enabling effective inhibition through sustained interactions rather than maximal static affinity.
Comparative analysis of the two sites indicates that ligands that favor binding at Site 1 tend to reduce protein flexibility and promote more coherent dynamic behavior, features commonly associated with functionally relevant inhibition of PTP1B. In this context, compounds 5a and 5d emerge as the most balanced ligands, combining acceptable binding energetics with favorable dynamic stabilization of the enzyme. Conversely, ligands primarily optimized for Site 1 binding appear to rely on stronger energetic contributions that are not necessarily accompanied by optimal dynamic stabilization.
Taken together, these results suggest that dynamic stabilization of PTP1B, rather than maximal binding free energy alone, is a key determinant of effective ligand behavior, and support Site 1 as a relevant target region for further optimization of this compound series. However, orthogonal experimental validation (e.g., mutagenesis, biophysical binding assays, or competition studies) will be required in future work to confirm the proposed interaction site.
To assess whether amino acid conjugates exhibit favorable drug-like properties, including pharmacokinetic properties, they were analyzed using the SwissADME server.? In Table S3 and Figures S33−S37 of the Supporting Information, the physicochemical properties are predicted (lipophilicity, size, polarity, solubility, flexibility, and saturation). Solubility is indicated by Log S, where insoluble (<−10), poorly (<−6), moderately (<−4), soluble (<−2), and highly (<0).
The predicted aqueous solubility shows a favorable trend when Gly (−9.21) or Arg (−9.22) is added at C30 of compound 4 (−9.48). On the other hand, unsaturation is determined by the fraction of carbons in the *sp^3^
- hybridization, which should be no less than 0.25. At the same time, that flexibility is assessed by the number of rotatable bonds, which should not exceed 9.? Compound 4 and derivatives 5a–5d demonstrated sp ^ 3 ^ fractions greater than 0.25. Notably, 5a (0.67) and 5b (0.65) showed lower values than compounds 4 (0.68) and 5c (0.68), respectively. Furthermore, TPSA is the sum of the contributions to the molecular (usually van der Waals) surface area of polar atoms such as oxygen, nitrogen, and their attached hydrogens; the values of TPSA in Å^2^ within the range 140 > TPSA > 60 are indications of excellent intestinal absorption and good blood-brain barrier penetration. All compounds demonstrated TPSA values >60 and <140 Å^2^. Additionally, the pharmacokinetic analysis predicted that compound 5b, but not compounds 4, 5a, 5c, and 5d, are substrates of the permeability glycoprotein (P-gp), an important protein in cell membranes that actively pumps many extraneous substances out of cells.? Despite the low absolute solubility values, the structural modifications significantly improved the polar surface area (TPSA). However, further experimental solubility assays and pharmacokinetic (PK) studies are necessary to validate these in silico predictions and confirm the impact of these structural modifications on the compounds’ bioavailability.
Based on these predictions and their favorable in vitro profiles, compounds 5a, 5b, and 5d were selected to assess their effects on GLUT4 expression and to conduct in vivo evaluations to further investigate their potential antihyperglycemic and insulin-sensitizing effects.
To assess whether inhibiting PTP1B with the new compounds enhances GLUT4 expression through increased insulin sensitivity, we evaluated GLUT4 levels in C2C12 myoblasts in the presence or absence of insulin (Figure). Pioglitazone, an insulin sensitizer and PPAR-γ agonist that enhances GLUT4 expression, ?−? ? was used as a positive control. In the absence of insulin, only Pioglitazone induced a significant GLUT4 expression. Interestingly, in the presence of insulin (0.8 μM), compounds 4, 5a, 5b, and 5d at 1 μM significantly increased GLUT4 expression, suggesting their potential to enhance insulin-stimulated glucose uptake. Moreover, despite being tested at concentrations five times lower, GLUT4 expression levels were comparable to those observed with pioglitazone.
Lastly, to evaluate the antihyperglycemic effect of compounds 4, 5a, 5b, and 5d, an oral glucose tolerance test (OGTT) was performed in nondiabetic mice (FigureA). CD-1 mice with STZ-induced diabetes are known to exhibit insulin resistance;? therefore, an insulin tolerance test (ITT) was conducted in this model to investigate the insulin-sensitizing effects of compounds 4, 5a, 5b, and 5d (FigureB). Pioglitazone was used as a positive control. The results of this study indicated that both pioglitazone and compound 5a significantly reduced the maximum blood glucose concentration in the OGTT compared to the nondiabetic control group. Although the effect of 5a was less pronounced than that of pioglitazone, the observed reduction indicates a moderate improvement in glucose tolerance, suggesting partial activation of insulin-sensitizing pathways.
In contrast, the ITT in STZ-diabetic mice showed that compound 4 (50 mg/kg) and its amino acid conjugate 5a (50 mg/kg) markedly decreased glucose levels, suggesting a positive impact on insulin sensitivity. Notably, compound 5b (50 mg/kg) reduced blood glucose levels comparable to those of pioglitazone (45 mg/kg) in this acute assay, highlighting its insulin-sensitizing potential. However, further investigations involving other in vivo insulin-resistant models are necessary.
Collectively, these findings provide a preliminary proof-of-concept that compound 5b, while exhibiting moderate antihyperglycemic activity under nondiabetic conditions, can exert a significant insulin-sensitizing effect in a diabetic model. The observed activity supports its potential as a promising lead candidate for further investigation as an insulin-sensitizing agent.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1IDF Diabetes Atlas: International Diabetes Federation: 2025; https://diabetesatlas.org (accessed 2025-09-29).
- 2Gan L.Chen P.Zhang Z.He X.Wu X.Chen Z.Wei Q.Wu Y.Static Training Improves Insulin Resistance in Skeletal Muscle of Type 2 Diabetic Mice via the IGF-2/IGF-1R Pathway Sci. Rep.20251511066210.1038/s 41598-025-94360-z 40148427 PMC 11950239 · doi ↗ · pubmed ↗
- 3Bragg F.Li L.Yang L.Guo Y.Chen Y.Bian Z.Chen J.Collins R.Peto R.Wang C.Dong C.Pan R.Zhou J.Xu X.Chen Z.Risks and Population Burden of Cardiovascular Diseases Associated with Diabetes in China: A Prospective Study of 0.5 Million Adults P Lo S Med.2016137 e 100202610.1371/journal.pmed.100202627379518 PMC 4933372 · doi ↗ · pubmed ↗
- 4Chaudhury A.Duvoor C.Reddy Dendi V. S.Kraleti S.Chada A.Ravilla R.Marco A.Shekhawat N. S.Montales M. T.Kuriakose K.Sasapu A.Beebe A.Patil N.Musham C. K.Lohani G. P.Mirza W.Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management Front. Endocrinol. (Lausanne).20178110.3389/fendo.2017.0000628144230 PMC 5239792 · doi ↗ · pubmed ↗
- 5Hossain, M. A. ; Pervin, R. Current Antidiabetic Drugs. In Nutritional and Therapeutic Interventions for Diabetes and Metabolic Syndrome; Elsevier: 2018; pp 455–473. 10.1016/B 978-0-12-812019-4.00034-9. · doi ↗
- 6Krishnan N.Koveal D.Miller D. H.Xue B.Akshinthala S. D.Kragelj J.Jensen M. R.Gauss C. M.Page R.Blackledge M.Muthuswamy S. K.Peti W.Tonks N. K.Targeting the Disordered C Terminus of PTP 1B with an Allosteric Inhibitor Nat. Chem. Biol.201410755856610.1038/nchembio.152824845231 PMC 4062594 · doi ↗ · pubmed ↗
- 7Liu R.Mathieu C.Berthelet J.Zhang W.Dupret J. M.Rodrigues Lima F.Human Protein Tyrosine Phosphatase 1B (PTP 1B): From Structure to Clinical Inhibitor Perspectives Int. J. Mol. Sci.20222313702710.3390/ijms 2313702735806030 PMC 9266911 · doi ↗ · pubmed ↗
- 8Mendoza-Jasso M. E.Pérez-Villanueva J.Alvarado-Rodríguez J. G.González-Andrade M.Cortés-Benítez F.3-Benzylaminomethyl Lithocholic Acid Derivatives Exhibited Potent and Selective Uncompetitive Inhibitory Activity Against Protein Tyrosine Phosphatase 1B (PTP 1B)ACS Omega 202410.1021/acsomega.4c 04948 · doi ↗