Left Inguino-Scrotal-Perineal Neurofibroma: A Report of a Rare Case and Review of Literature
Riyadh Alloqmani, Yazeid I Alrefaey, Mohammed Alghamdi, Jana Hareesi, Salahaldin Lamy, Abeer Nafadi, Abdullah Qashgry

TL;DR
A rare case of a neurofibroma in the left inguino-scrotal area is reported, highlighting its diagnosis and treatment.
Contribution
This paper presents a rare case of paratesticular neurofibroma and emphasizes its inclusion in differential diagnosis.
Findings
Neurofibroma was confirmed via histopathology and immunohistochemistry in a 66-year-old patient.
Complete surgical excision is recommended as the treatment for paratesticular neurofibroma.
Abstract
Paratesticular masses arise from structures surrounding the testes, including the spermatic cord, epididymis, vestigial remnants, and tunica vaginalis. These masses are often benign, such as lipomas, leiomyomas, and adenomatoid tumors. Neurofibroma (NF) is a benign tumor of Schwann cell origin, characterized by strong S-100 protein expression. Neurofibromas typically occur in the face, shoulders, arms, periungual regions, and feet; the involvement of the inguino-scrotal region is extremely rare. We report a case of a 66-year-old man with a left inguino-scrotal mass of three years’ duration that progressively enlarged and became painful. After a multidisciplinary review, surgical excision was performed. Histopathology and immunohistochemistry confirmed paratesticular neurofibroma. Neurofibroma should be considered in the differential diagnosis of paratesticular masses, and complete…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
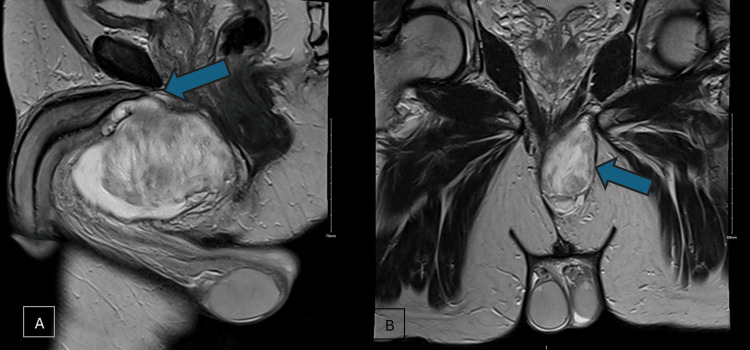 Figure 1
Figure 1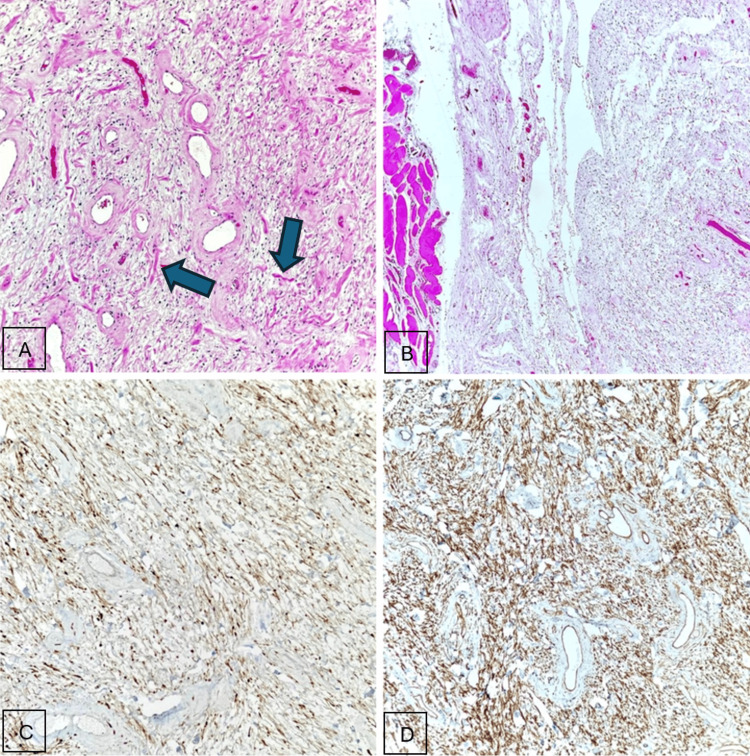 Figure 2
Figure 2| Year | Author | Age | Location | Subtype | Tumor Size (cm) | Imaging Findings | Management | Outcome |
| 1939 | Schulte et al. [ | 49 | Spermatic cord | Typical | Not reported | Not reported | Radical orchiectomy | Not reported |
| 1948 | Levant and Chetlin [ | 54 | Intratesticular (tunica albuginea) | Typical | Not reported | Not reported | Simple orchiectomy | No recurrence |
| 1975 | Jepson [ | 21 | Extratesticular (multiple lesions) | Typical | Not reported | Not reported | Debulking (partial excision) | Not reported |
| 1977 | Livolsi and Schiff [ | 23 | Intratesticular | Typical | Not reported | Not reported | Radical orchiectomy | No recurrence |
| 1982 | Yamamoto et al. [ | 8 | Extratesticular | Typical | 5.0 × 1.5 × 1.5 | Not reported | Complete excision (testis-sparing) | No recurrence (one year) |
| 1990 | Yoshimura et al. [ | 41 | Extratesticular | Typical | 3.0 × 3.5 × 5.0 | Not reported | Complete excision (testis-sparing) | No recurrence (one year) |
| 1993 | Issa et al. [ | 77 | Extratesticular | Typical | 15 × 4.5 × 3.0 | Ultrasound (US): solid scrotal mass | Complete excision (testis-sparing) | No recurrence (one year) |
| 2002 | Deliveliotis et al. [ | 74 | Spermatic cord | Typical | 4.0 × 4.0 × 1.0 | Not reported | Radical orchiectomy | No recurrence (four months) |
| 2004 | Milathianakis et al. [ | 86 | Spermatic cord | Typical | Not reported | Not reported | Complete excision (testis-sparing) | No recurrence (19 months) |
| 2004 | Türkyilmaz et al. [ | 11 | Extratesticular | Typical | Not reported | Not reported | Complete excision (testis-sparing) | No recurrence (one year) |
| 2008 | Erdemir et al. [ | 45 | Extratesticular | Typical | 3.0 × 2.0 × 1.0 | US: hypoechoic; CT: ~3 cm mass | Complete excision (testis-sparing) | No recurrence |
| 2012 | Hosseini et al. [ | 45 | Extratesticular | Typical | 9.0 × 4.5 | US: hypoechoic; CT: 9 cm mass | Complete excision (testis-sparing) | No recurrence (five months) |
| 2015 | Boto et al. [ | 38 | Spermatic cord | Typical | ≈3.0-4.0 | MRI: T2 hyperintense mass | Complete excision (testis-sparing) | No recurrence |
| 2016 | Chandrashekar et al. [ | 32 | Extratesticular (inguino-scrotal) | Typical | 10 × 20 | Not reported | Complete excision (testis-sparing) | Not reported |
| 2021 | Thorat and Khachane [ | 77 | Extratesticular | Typical | 20 × 9.0 | US: heterogeneous hypoechoic mass | Complete excision (testis-sparing) | No recurrence (one month) |
| 2024 | Jaber et al. [ | 6 | Extratesticular | Plexiform | ≈5.5 × 3.0 | MRI: T2 hyperintense; target sign | Complete excision (testis-sparing) | No recurrence (one year) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTesticular diseases and treatments · Neuroblastoma Research and Treatments · Adrenal and Paraganglionic Tumors
Introduction
Neurofibromas (NF) are benign peripheral nerve sheath tumors that may occur as solitary lesions or in association with neurofibromatosis type 1 (NF1), caused by the mutation of the NF1 tumor-suppressor gene on chromosome 17q11.2 [1]. They are composed of Schwann cells, fibroblasts, perineural cells, and mast cells. Neurofibromas may be localized, diffuse, or plexiform. Solitary neurofibromas can arise throughout the body, most commonly in the neck, thorax, retroperitoneum, and extremities. Paratesticular involvement is exceptionally rare [2-4].
Molecular testing has a limited diagnostic role. Copy number analysis may assist in evaluating atypical neurofibroma or distinguishing it from malignant peripheral nerve sheath tumor (MPNST), which typically demonstrates a complex genomic profile [1].
Most paratesticular masses originate from the epididymis, spermatic cord, or tunica vaginalis and are typically benign. Given the rarity of paratesticular neurofibromas and their nonspecific clinical and imaging features, accurate preoperative diagnosis is challenging, making surgical exploration and definitive histopathological assessment essential for both diagnosis and appropriate management, thereby highlighting the clinical importance of this case.
Case presentation
A 66-year-old man presented with a left inguino-scrotal mass present for three years, which had progressively enlarged over the past year and recently became painful. The patient denied constitutional symptoms, trauma, prior scrotal surgery, urinary symptoms, flank pain, or hematuria. Physical examination revealed a rubbery, well-circumscribed mass extending from the upper left scrotum to the scrotal neck; both testes were normal and separable.
Pelvic MRI demonstrated a large, elongated left perineal mass measuring 10.9 × 4.1 × 7.2 cm with heterogeneous peripheral enhancement. The mass was inseparable from the root of the penis, exerting mass effect on the proximal urethra and adjacent structures, prompting a broad differential diagnosis (Figure 1) [5,6].
MRI of the scrotum post gadolinium.(A) Sagittal view of the large perineal mass with heterogeneous peripheral enhancement, which is inseparable from the root of the penis (arrowhead). (B) Coronal view of the mass, away from the testis (arrowhead).
A multidisciplinary tumor board recommended surgical removal, with spermatic cord liposarcoma considered the leading preoperative differential. A longitudinal left inguino-scrotal incision was made, and the mass was excised en bloc while preserving the spermatic cord and adjacent structures.
Gross evaluation revealed a specimen measuring 11.5 × 7.5 × 3.5 cm with a smooth external surface and a heterogeneous tan-yellow cut surface with focal hemorrhage.
Microscopically, the tumor consisted of spindle cells with bland, wavy nuclei and collagen fibrils. Mast cells were readily identifiable, and blood vessels showed hyalinized walls. No Verocay bodies, necrosis, atypical mitoses, or high-grade features were observed. Immunohistochemistry demonstrated strong S-100 positivity and diffuse CD34 expression. Tumor cells were negative for SOX10, SMA, desmin, caldesmon, myogenin, MUC4, CK-pan, calponin, EMA, STAT6, and PR (Figure 2) [1,4,7-9].
Histologic photomicrographs of the lesion showing (H&E: A, 400×; C, 40×) a spindle-cell neoplasm associated with a variably myxoid to collagenous stroma. Stromal collagen has a characteristic appearance resembling shredded carrots (arrowheads). (B) Neoplastic Schwann cells are represented by bland, spindle-shaped cells with thin, wavy nuclei, highlighted by S-100. (D) Classically, CD34 shows a strong and diffuse latticelike expression pattern in neurofibromas.
The postoperative course was uneventful. At the six-month follow-up, there was no evidence of recurrence.
Discussion
This case highlights the importance of distinguishing neurofibroma from other paratesticular soft-tissue tumors. Differential considerations include liposarcoma, cellular angiofibroma, and schwannoma. In this case, the immunoprofile, strong S-100 positivity and CD34 expression, with the absence of SOX10, desmin, SMA, EMA, and STAT6, supported the diagnosis of neurofibroma.
Paratesticular neurofibroma is exceptionally rare, with fewer than 20 cases reported in the English literature [4,7-12]. Patients typically present with a painless or mildly painful scrotal mass. MRI provides valuable information regarding lesion size, extent, and the involvement of surrounding structures, facilitating surgical planning.
Histologically, neurofibromas are unencapsulated spindle-cell lesions that are S-100-positive [1,7,8,10-12]. Although SOX10 negativity is uncommon, the combination of morphology and S-100 expression remains diagnostic. Complete surgical excision is curative and carries a low risk of recurrence. Evaluation for NF1 should be considered when there are multiple lesions or systemic features, including café-au-lait spots, Lisch nodules, sphenoidal dysplasia, and optic gliomas [1,7,9].
At the molecular level, most neurofibromas are driven by the inactivation of the NF1 tumor-suppressor gene on chromosome 17q11.2, which encodes neurofibromin, a negative regulator of RAS-MAPK signaling [1]. The loss of NF1 function leads to the constitutive activation of the RAS pathway, promoting Schwann cell proliferation.
However, molecular testing is rarely performed in neurofibromas. Many published genital cases, including scrotal, spermatic cord, and tunica albuginea neurofibromas, report no clinical signs of NF1 and rely solely on histology and immunohistochemistry for diagnosis (Table 1) [7,9,12-14]. Pediatric cases have also shown classic morphology without the genetic evidence of NF1 [11].
The WHO classification notes that molecular testing is not required for diagnosing conventional neurofibroma but may be helpful in atypical lesions. Atypical neurofibromas often show CDKN2A/CDKN2B deletions, while MPNSTs exhibit complex genomic alterations “including TP53 and PRC2 component loss” [1]. Older reports of genital neurofibroma predated modern molecular testing, but authors occasionally noted that cytogenetic studies may help differentiate neurofibroma from low-grade sarcoma [7].
Malignant transformation is exceedingly rare in solitary sporadic neurofibroma and is primarily associated with plexiform neurofibroma in patients with NF1. No malignant transformation has been reported in genital neurofibromas without NF1 [8-12]. For tumors with benign morphology, low mitotic activity, and classic immunophenotype, routine molecular analysis is unlikely to alter management. Nonetheless, in large, rapidly growing, or histologically atypical paratesticular tumors, targeted molecular studies (NF1, CDKN2A/CDKN2B, and copy number profiling) may help exclude early MPNST.
In our patient, the benign histologic features, conventional immunoprofile, absence of NF1 stigmata, and uneventful postoperative course strongly support a diagnosis of a sporadic neurofibroma, with no indication for additional molecular workup.
Conclusions
Paratesticular neurofibroma is a rare benign tumor requiring thorough clinical, radiologic, and histopathological assessment to ensure accurate diagnosis. Complete surgical excision offers an optimal outcome, and short-term follow-up is recommended to monitor for recurrence. Although recurrence is extremely rare, routine long-term surveillance is not mandated by the WHO criteria. The awareness of this rare entity helps prevent misdiagnosis and avoid unnecessary radical surgical interventions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1The 2020 WHO classification of soft tissue tumours: news and perspectives Pathologica Sbaraglia M Bellan E Dei Tos AP 708411320213317961410.32074/1591-951X-213PMC 8167394 · doi ↗ · pubmed ↗
- 2Extratesticular intraescrotal neurofibroma: case report (Article in Spanish)Arch Esp Urol Trovarelli S Tallis V Tripodi S Miracco C Ponchietti R 905908592006 https://scielo.isciii.es/scielo.php?pid=S 0004-06142006000900010&script=sci_arttext&utm_source=chatgpt.com 1719021510.4321/s 0004-06142006000900010 · doi ↗ · pubmed ↗
- 3Solitary neurofibroma of scrotum J Urol Yoshimura K Maeda O Saiki S Kuroda M Miki T Usami M Kotake T 8231431990231381810.1016/s 0022-5347(17)40109-1 · doi ↗ · pubmed ↗
- 4Solitary neurofibroma of the spermatic cord Int Urol Nephrol Deliveliotis C Albanis S Skolarikos A Varkarakis J Protogerou V Tamvakis N Alargof E 3733753420021289923110.1023/a:1024415016017 · doi ↗ · pubmed ↗
- 5Multimodality imaging of paratesticular neoplasms and their rare mimics Radiographics Akbar SA Sayyed TA Jafri SZ Hasteh F Neill JS 146114762320031461555810.1148/rg.236025174 · doi ↗ · pubmed ↗
- 6Adult paratesticular tumours BJU Int Khoubehi B Mishra V Ali M Motiwala H Karim O 7077159020021241075310.1046/j.1464-410x.2002.02992.x · doi ↗ · pubmed ↗
- 7Intrascrotal extratesticular neurofibroma Urology Yamamoto M Miyake K Mitsuya H 200201201982711283410.1016/0090-4295(82)90364-8 · doi ↗ · pubmed ↗
- 8Intrascrotal neurofibromas Urology Issa MM Yagol R Tsang D 350352411993847032210.1016/0090-4295(93)90594-z · doi ↗ · pubmed ↗