Prevalence of actionable pharmacogenomic variants in Brazilian patients with cancer
Jaqueline B. Schuch, Mariana R. Botton, Angélica C. De Baumont, Giovana Curzel, Nathan A. Cadore, Cláudia Bordignon, Mahira L. Rosa, Vitor F. Vasconcellos, Lilian A. R. Barros, Cristiano P. Souza, Williams F. Barra, Daniela L. C. Louzeiro, Alessandra Notari

TL;DR
This study found that nearly all Brazilian cancer patients have genetic variants that could affect drug response, suggesting the importance of personalized medicine in oncology.
Contribution
The study provides the first detailed analysis of actionable pharmacogenomic variants in Brazilian cancer patients.
Findings
99.33% of participants had at least one actionable pharmacogenomic variant.
Common variants included VKORC1 rs9923231 T (32.63%) and ABCG2 rs2231142 T (8.96%).
UGT1A1 had decreased-function alleles in 37% of participants.
Abstract
Pharmacogenomic (PGx) variants can influence drug efficacy and safety, yet their prevalence in Latin American populations with cancer is underexplored. Our aim is to characterize the frequency and phenotypic distribution of actionable pharmacogenes in Brazilian patients with metastatic prostate cancer (MPC) and Human Epidermal Growth Factor Receptor 2 (HER2)-positive breast cancer (BC). This analysis included 452 patients (259 BC, 193 MPC) from a multicenter study across 19 Brazilian sites. Exome sequencing was performed, and PGx variants were analyzed using the Pharmacogenomics Clinical Annotation Tool (PharmCAT) following the Clinical Pharmacogenetics Implementation Consortium (CPIC®) guidelines. Genotypes, star alleles, and predicted phenotypes were reported for 15 clinically relevant pharmacogenes. Actionable PGx phenotypes were detected in 99.33% of participants. The…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
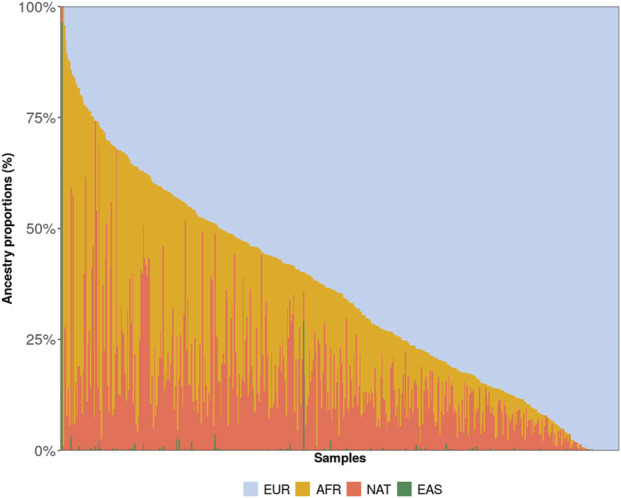 FIGURE 1
FIGURE 1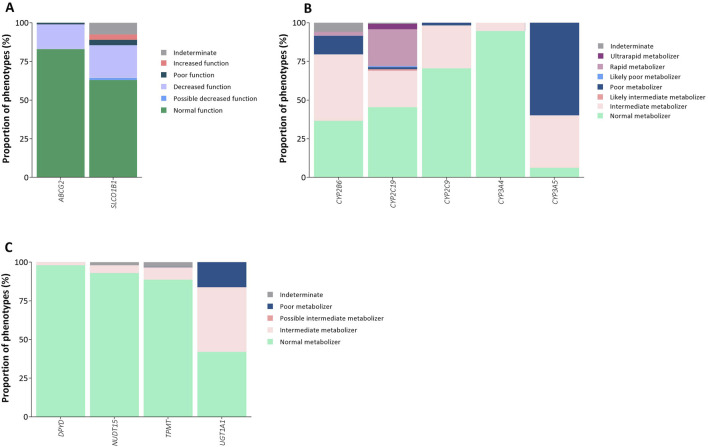 FIGURE 2
FIGURE 2| Variable | Women with HER2+ BC (n = 259) | Men with MPC (n = 193) |
|---|---|---|
| Age (years), mean (SD) | 52.2 (11.69) | 68.4 (9.50) |
| Ethnicity* | ||
| White | 120 (49.38) | 98 (50.78) |
| Black | 32 (13.17) | 22 (11.40) |
| Mixed | 89 (36.63) | 72 (37.31) |
| Asian | 2 (0.82) | 1 (0.52) |
| Brazilian macro-region of birth | ||
| Central-west | 16 (6.23) | 11 (5.70) |
| Northeast | 46 (17.90) | 58 (30.05) |
| North | 24 (9.34) | 15 (7.77) |
| Southeast | 106 (41.25) | 52 (26.94) |
| South | 65 (25.29) | 57 (29.53) |
| Clinical comorbidities | 120 (48.00) | 118 (61.78) |
| Systemic arterial hypertension | 66 (25.48) | 97 (50.26) |
| Diabetes mellitus | 32 (12.36) | 33 (17.10) |
| Ischemic heart disease | 3 (1.16) | 11 (5.70) |
| Dyslipidemias | 12 (4.63) | 13 (6.74) |
| Hypothyroidism | 22 (8.49) | 5 (2.59) |
| Gene | Protein change | Variant ID | Allele frequency | SIFT | PolyPhen |
|---|---|---|---|---|---|
|
| p.Ile237Val | rs1007241293 | 0.11% | Tolerated | Benign |
| | p.Gly256Arg | rs754247932 | 0.11% | Deleterious | Probably damaging |
| | p.Gln327His | rs151155254 | 0.11% | Tolerated | Benign |
|
| p.Ser173Cys | rs148377536 | 0.33% | Tolerated | Benign |
| | p.Ile209Val | rs144518874 | 0.33% | Tolerated | Benign |
| | p.Glu339Ala | rs565104467 | 0.33% | Tolerated | Benign |
| | p.His341Asp | rs138030127 | 0.33% | Tolerated | Benign |
| | p.Val367Leu | rs143979776 | 0.33% | Tolerated | Benign |
| | p.Arg323Gly | rs201282330 | 0.11% | Deleterious | Probably damaging |
| | p.Thr67Met | rs138264188 | 0.44% | Deleterious | Probably damaging |
| | p.Lys91Asn | rs772100005 | 0.22% | Tolerated | Benign |
| | p.Asp257Asn | rs34646544 | 0.11% | Deleterious | Benign |
| | p.Asp266Asn | rs770007043 | 0.11% | Deleterious | Benign |
|
| p.Asn474Ser | rs1031294281 | 0.55% | Tolerated | Benign |
| | p.Thr130Met | rs150152656 | 0.11% | Deleterious | Probably damaging |
| | p.Val113Ile | rs145119820 | 0.22% | Tolerated | Benign |
| | p.Ile60Thr | rs1848387737 | 0.11% | Tolerated | Benign |
| | p.Glu81Lys | rs149072229 | 0.11% | Tolerated | Benign |
| | p.Ile88Thr | rs1471083174 | 0.11% | Deleterious | Benign |
| | p.Arg125His | rs141774245 | 0.11% | Tolerated | Benign |
| | p.Arg132Trp | rs149590953 | 0.11% | Deleterious | Probably damaging |
| | p.Ser336Ile | rs143833145 | 0.11% | Deleterious | Benign |
| | p.Leu380Val | rs1364388853 | 0.11% | Tolerated | Benign |
| | p.Asn423Lys | rs758626485 | 0.11% | Deleterious | Benign |
|
| p.Glu104Asp | rs367848139 | 0.11% | Tolerated | Benign |
| | p.Arg329Cys | rs768830601 | 0.11% | Tolerated | Possibly damaging |
| | p.Ile389Thr | rs753971542 | 0.11% | Tolerated | Benign |
| | p.His410Arg | rs746440122 | 0.11% | Deleterious | Benign |
|
| p.Arg403Cys | rs143966082 | 0.22% | Deleterious | Probably damaging |
| | p.Met395Val | rs142425279 | 0.11% | Tolerated | Benign |
| | p.Thr310Lys | rs751246524 | 0.11% | Deleterious | Benign |
|
| p.Asn247His | rs1810716398 | 0.11% | Deleterious | Benign |
| | p.Ile149Thr | rs142823108 | 0.11% | Deleterious | Probably damaging |
| | p.Ser100Tyr | rs41279857 | 0.22% | Deleterious | Probably damaging |
|
| p.Ala483Gly | rs3952537 | 4.95% | Deleterious | Benign |
| | p.Leu514Pro | rs777992718 | 0.11% | Deleterious | Probably damaging |
| | p.Ser423Asn | rs780963140 | 0.22% | Tolerated | Benign |
| | p.Ile406Thr | rs754740791 | 0.11% | Tolerated | Benign |
| | p.His401Arg | rs1435072497 | 0.11% | Tolerated | Benign |
| | p.Arg400Cys | rs75222722 | 0.22% | Deleterious | Probably damaging |
| | p.Glu308Gly | rs200477560 | 0.11% | Deleterious | Probably damaging |
| | p.Arg276Cys | rs61739998 | 0.11% | Tolerated | Benign |
| | p.Asp265Asn | rs750598459 | 0.11% | Tolerated | Benign |
| | p.Lys161Arg | rs138811366 | 0.33% | Deleterious | Possibly damaging |
| | p.Ser2Phe | rs144146357 | 0.22% | Tolerated | Benign |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPharmacogenetics and Drug Metabolism · Drug Transport and Resistance Mechanisms · Histone Deacetylase Inhibitors Research
Introduction
1
Pharmacogenomics (PGx) involves the understanding of individual responses to drug therapies, including efficacy and toxicity, by analyzing the effects of genetic variability on the metabolism of drugs and prodrugs (Relling and Evans, 2015). This individual variability is of major clinical importance due to its potential association with drug efficacy and adverse drug reactions, ultimately affecting patient care and causing substantial health issues. Large-scale genotyping studies have shown that nearly all individuals carry at least one PGx variant that could impact outcomes related to medication use (Bush et al., 2016; Ji et al., 2016; Lanillos et al., 2022; McInnes et al., 2021; Van Driest et al., 2014).
Drug-response phenotypes can be partly attributed to individual genetic variability. A well-established example involves dihydropyrimidine dehydrogenase (DPYD) variants, which affect the metabolism of fluoropyrimidines in patients with colorectal cancer. Pathogenic variants in this gene are strongly associated with an increased risk of potentially life-threatening toxicity during treatment with agents such as 5-fluorouracil or capecitabine (Amstutz et al., 2018; NCCN, 2025). For this reason, international guidelines, including those from the National Comprehensive Cancer Network (NCCN), recommend DPYD testing in specific clinical settings. Other clinically relevant PGx applications include the assessment of uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) polymorphisms, associated with irinotecan-induced toxicity, and thiopurine S-methyltransferase (TPMT) and nudix hydrolase 15 (NUDT15) variants, predictive of adverse reactions to thiopurines used in certain hematologic malignancies (Gammal et al., 2016; Relling et al., 2019). These advances illustrate how the integration of PGx into cancer care can optimize therapeutic efficacy while improving patient safety (NCCN, 2025; Varughese et al., 2020).
Several challenges hinder the implementation of PGx, among which extraction and analysis of genomic variants are major obstacles (Klein and Ritchie, 2018). For instance, the cytochrome P450 (CYP) gene family is the most important gene family in PGx, as CYP genes are highly polymorphic. These genes encode cytochrome P450 proteins and are relevant in 10%–20% of all drug therapies (Zhou and Lauschke, 2022). To address the challenges in PGx assessment, specific PGx tools have been developed to identify PGx genotypes, infer haplotypes, and predict phenotypes. One such tool is the Pharmacogenomics Clinical Annotation Tool (PharmCAT), which adheres to the most recent PGx guidelines and uses a predictive framework to determine haplotypes in 18 actionable pharmacogenes, as well as the associated phenotypes (Klein and Ritchie, 2018).
Population admixture adds further complexity to the implementation of PGx. In admixed populations, the absence of comparable groups with relatively homogeneous genetic ancestry complicates the interpretation of results and hinders the implementation of genomic medicine into clinical practice, requiring specific genomic studies of these individuals. The genetic composition of the Brazilian population, with ancestry contributions from African, Native American, and European populations, has likely contributed to a genetically admixed population that remains underrepresented in public genomic reference databases (Mychaleckyj et al., 2017; Rodrigues De Moura et al., 2015; Secolin et al., 2019). A recent large-scale genomic study of the Brazilian population identified more than 8 million previously unreported genetic variants, highlighting both its extensive genetic diversity and the significant underrepresentation of admixed populations in global genomic databases (Nunes et al., 2025). Another study of individuals from the Southeast of Brazil investigated 38 pharmacogenes and found that 98% of participants carried at least one high-risk genotype-predicted phenotype, although a search for novel alleles was not included (Bertholim-Nasciben et al., 2023).
In the present study, we investigated the genetic frequency and phenotypic variability of actionable pharmacogenes in the Brazilian population. We used exome sequencing to identify variants in protein-coding pharmacogenes, enabling the large-scale study of variants, in men with metastatic prostate cancer (MPC) and women with Human Epidermal Growth Factor Receptor 2 (HER2)-positive breast cancer (BC). PGx variant selection was analyzed and interpreted according to the Clinical Pharmacogenetics Implementation Consortium (CPIC®) guidelines and implemented using PharmCAT, based on evidence from the Clinical Pharmacogenomic (ClinPGx) database and genomic data.
Materials and methods
2
Study design and participants
2.1
This is a secondary analysis of data derived from Onco-Genomas Brasil, a multicenter study conducted in 19 health facilities distributed among the 5 macro-regions of Brazil, ensuring broad geographic representation across the national territory. The sample size was calculated to estimate the prevalence of germline genetic alterations in cancer predisposition genes and was not powered for the detection of rare variants. Therefore, rare variants identified in this study are reported descriptively and should be interpreted as exploratory findings. The Onco-Genomas Brasil study is part of the Brazilian Genome Program (Programa Genomas Brasil) of the Ministry of Health (Ministério da Saúde). All participants received care in public hospitals through the Brazilian Unified Health System (Sistema Único de Saúde - SUS).
The sample consisted of 259 women with locally advanced HER2-positive BC and 193 men with MPC. The inclusion criteria for women with BC were age ≥18 years, histologically confirmed breast carcinoma with HER-2 overexpression (a score of 3+ or 2+ on immunohistochemistry with positive in situ hybridization), clinical stage II or III according to the American Joint Committee on Cancer (AJCC) classification, and use of neoadjuvant treatment with chemotherapy plus trastuzumab. For men with MPC, the inclusion criteria were age ≥18 years, histologically confirmed prostate adenocarcinoma, and clinical stage IV according to the AJCC classification.
Demographic data were collected from participant interviews and/or medical records. Ethno-racial self-identification was obtained through interviews and classified according to the categories defined by the Brazilian Institute of Geography and Statistics (IBGE): (1) White; (2) Black; (3) Mixed; (4) Asian; and (5) Indigenous. The detailed study protocol has been published previously (Schuch et al., 2024). Descriptive data for continuous variables are expressed as mean and standard deviation (SD) or median and interquartile range (IQR), depending on the data distribution. Categorical data are expressed as absolute and relative frequencies.
The study protocol was registered at ClinicalTrials.gov (NCT05306600) and approved by the research ethics committee of the coordinating center, Hospital Moinhos de Vento (CAAE 55457122.3.1001.5330), and by the local ethics committees of all participating centers. Written informed consent was obtained from all participants before their inclusion in the study.
Sample processing
2.2
Peripheral blood samples were collected from each participant and transported to the coordinating center for processing. Germline deoxyribonucleic acid (DNA) was extracted using the QIAamp DNA Blood Kit (Qiagen). The minimum sample quality parameters were defined as an absorbance ratio of 260:280 between 1.8 and 2.0 (NanoDrop) and a minimum total yield of 250 nanogram (ng), quantified with the Qubit™ 1X dsDNA High Sensitivity Kit (Invitrogen™, ref Q33231) using the Qubit™ 4 Fluorimeter (Invitrogen™, ref Q33238). These criteria were adopted to optimize the performance of exome sequencing.
Next-generation sequencing (NGS) was performed using the Illumina NovaSeq 6,000 platform. Library preparation was conducted with the Twist Library Preparation EF 2.0 Kit, target enrichment with the Twist Target Enrichment Standard Hybridization v2 Kit, and sequencing with the NovaSeq 6000 S4 Reagent Kit. The quality parameters for NGS included an average size of the DNA library obtained by Tapestation between 375 and 450 bp, and a minimum size of the sequenced target DNA fragment between 100 and 150 bp. Each sample achieved at least 50X mean coverage, a minimum Q30 of 85%, and contamination levels below 2%. Across all samples included in the analysis, the proportion of bases covered at >20X was 98.7% ± 0.08%, and the proportion of bases with Q30 or higher was 91.2% ± 1.4%. Sequence alignment and mapping were performed using Dynamic Read Analysis for GENomics (DRAGEN) v.3.10.4, using the Genome Reference Consortium Human Build 38 (GRCh38). Variant calling and quality filtering were performed using the DRAGEN pipeline with hard-filtering. The DRAGEN Joint Genotyping pipeline was used to merge and perform joint genotype calling of each sample into a multi-sample variant call format (VCF) file containing data from all germline samples and approximately 488,000 variants. Only variants marked as PASS after DRAGEN’s standard hard-filtering and joint genotyping were retained for pharmacogenomic analyses. The inference of the haplotypic phase from genomic data was obtained with Segmented HAPlotype Estimation and Imputation Tools version 5 (ShapeIT5) using the phase_common option, employing a cohort-based phasing framework without the use of an external reference panel, and the genetics maps in b38 for whole-genome sequencing chunks supplied by the tool (Hofmeister et al., 2023).
Pharmacogenomic analyses
2.3
The PharmCAT software was used to extract relevant PGx variants, infer diplotypes/genotypes, and predict phenotypes according to the CPIC® recommendations (Klein and Ritchie, 2018). The GRCh38 assembly was used as a reference. For each germline sample, a report was generated comprising genotype, allele functionality, and phenotype for the following genes: ATP Binding Cassette Subfamily G Member 2 (ABCG2), Interferon Lambda 3/4 (IFNL3/4), Vitamin K Epoxide Reductase Complex Subunit 1 (VKORC1), Solute Carrier Organic Anion Transporter Family Member 1B1 (SLCO1B1), Cytochrome P450 family genes (CYP2B6, CYP2C19, CYP2C9, CYP3A4, CYP3A5, CYP4F2), NUDT15, TPMT, UGT1A1, DPYD, and Ryanodine Receptor 1 (RYR1). Cytochrome P450 Family 2 Subfamily D Member 6 (CYP2D6) was not assessed due to the complexity of accurately detecting copy number variations in this gene. Consistent with PharmCAT recommendations, CYP2D6 genotyping using WES is not supported in the standard pipeline because of the presence of highly homologous pseudogenes and frequent structural rearrangements, requiring specialized analytical approaches or long-read sequencing for reliable characterization (Sangkuhl et al., 2020). If multiple genotype combinations were generated for the positions of interest, additional analyses were performed. The 15 pharmacogenes included in this analysis were selected based on their clinical relevance and actionability, guided by recommendations from established pharmacogenomic guidelines (e.g., CPIC and Dutch Pharmacogenetics Working Group - DPWG) and evidence curated in pharmacogenomic databases such as ClinPGx. These genes harbor well-characterized functional variants with strong levels of evidence and validated genotype–phenotype relationships, supporting their potential clinical implementation across multiple therapeutic areas. A complete list of variants analyzed using this approach is provided in Supplementary Table S1. VCF files were analyzed to identify previously unreported alleles not yet included in the Pharmacogene Variation (PharmVar) database (Gaedigk et al., 2018). Key pharmacogenetic non-coding variants, including regulatory and promoter markers, were adequately covered with high genotype quality through off-target capture; their respective average coverage and call rates are provided in Supplementary Table S2.
Uncertain diplotype calls were evaluated on a case-by-case basis following predefined decision criteria. Visual inspection of genomic data was performed using the Integrative Genomics Viewer (IGV) software, based on Binary Alignment/Map (BAM) files and their corresponding index (BAI) files for each sequenced sample. Variant positions and star-allele definitions were obtained from the PharmVar database (https://www.pharmvar.org/). When diplotype ambiguity could not be resolved through read-level inspection or when no predefined star-allele definition matched the observed variant pattern, the sample was excluded from the analysis for that specific gene. The number of samples excluded due to unresolved or unmatched diplotype calls was 3 for SLCO1B1, 8 for CYP2B6, 1 for CYP2C9, 1 for CYP3A5, and 34 for CYP4F2.
The classification of actionable phenotypes of interest included poor function for ABCG2; decreased and poor functions for SLCO1B1; presence of pathogenic or likely pathogenic variants for RYR1; poor, intermediate, rapid and ultrarapid metabolizers for CYP2B6 and CYP2C19; poor and intermediate metabolizers for CYP2C9, CYP3A5, DPYD, NUDT15, TPMT and UGT1A1; and poor metabolizers for CYP3A4.
Global ancestry inference
2.4
Global ancestry was inferred using the ADMIXTURE (Alexander et al., 2009) software by a supervised analysis for k = 4. To generate the reference panel, the multi-sample VCF file containing all target samples was merged with samples from the 1000 Genomes Project (Phase 3) for African, East Asian, and European populations and from the Human Genome Diversity Project (HGDP) for Native American continental populations (Bergström et al., 2020; Fairley et al., 2020). Hardy-Weinberg equilibrium (HWE)-normalized principal component analysis (PCA) (hwe_normalized_pca) was performed using Hail (hail.is) (Hail Team, 2025).
Results
3
Table 1 presents the demographic characteristics of the sample. Approximately half of the participants self-identified as White, and approximately 60% were born in the Southeast or South regions of Brazil. The median age at diagnosis was 64.47 (±9.60) years in men and 48 (IQR 41–57) years in women. Global ancestry inference revealed that the cohort was predominantly of European ancestry (64.32%, SD 23.63%), followed by African (20.60%, SD 18.79%) and Native American (14.69%, SD 12.58%) proportions, with East Asian ancestry representing <1% (Figure 1).
Genetic ancestry proportions estimated for all participants included in the study. NAT = Native American; AFR = African; EAS = East Asian; EUR = European.
The prevalence of actionable PGx phenotypes in our sample was 99.33%. Only 1 woman with BC and 2 men with MPC exhibit no actionable phenotypes. Among participants with actionable phenotypes, the majority carried 2 (n = 124, 27.43%) or 3 (n = 144, 31.86%) phenotypes, with a maximum of 6 (n = 3, 0.66%). Allelic and genotypic prevalences, along with their respective phenotypes, are presented in Supplementary Tables S3, S4.
A single variant was analyzed in 3 genes. The minor allele frequency of the ABCG2-rs2231142 T allele was 8.96%, which is associated with decreased function. Decreased and poor functions accounted for 16.15% (G/T genotype, n = 73) and 0.89% (T/T genotype, n = 4) of the predicted phenotypes, respectively (Figure 2A). The IFNL3/4-rs12979860 T-allele and the VKORC1-rs9923231 T-allele had frequencies of 40.82% and 32.63%, respectively.
Actionable phenotype prevalence in Brazilian patients with PC and BC. (A) Drug transporter genes, (B) Cytochrome P450 (CYP) genes, (C) Other Drug-metabolizing enzyme genes. Stacked bar plots show the proportional distribution of phenotype categories for each gene, with percentages summing to 100% within each bar. Sample sizes were as follows: ABCG2, DPYD, CYP2C19, NUDT15, TPMT, UGT1A1 (n = 452); CYP3A5, CYP2C9 (n = 451); SLCO1B1 (n = 450); CYP3A4 (n = 446); CYP2B6 (n = 444). Detailed counts of each phenotype category within each gene are provided in Supplementary Material.
A total of 17 SLCO1B1 alleles were identified in this Brazilian sample (Supplementary Table S3), and normal function was the most frequent phenotype (n = 284, 63.11%). The prevalence of decreased function was 21.11% (n = 95), with a lower prevalence of increased function (n = 16, 3.56%) and poor function (n = 16, 3.56%) (Figure 2A; Supplementary Table S4). The most common star allele was the normal function *SLCO1B1**37 (n = 179, 19.93%). Other observed alleles included the no function *15 (n = 116, 12.92%), and the increased function *14 (n = 93, 10.36%) and *20 (n = 58, 6.46%). Three missense variants in SLCO1B1 are not currently included among PharmVar-reported alleles (Table 2). Each variant was rare, identified in a single individual, corresponding to a frequency of 0.11%.
Across CYP genes, intermediate metabolizer was the most prevalent phenotype in CYP2B6 (n = 191, 43.02%), followed by the normal metabolizer (n = 162, 36.49%, Figure 2B). The reference *1 (n = 466, 52.48%) and the decreased function *6 (n = 254, 28.60%) alleles were the most common among the 19 star alleles identified in the sample. Five missense variants not included among PharmVar alleles were reported in the same three individuals with an allele frequency of 0.33%: rs148377536 (p.Ser173Cys), rs144518874 (p.Ile209Val), rs565104467 (p.Glu339Ala), rs138030127 (p.His341Asp), and rs143979776 (p.Val367Leu). Another unreported missense variant, rs201282330 (p.Arg323Gly), was found in one individual. Importantly, this individual was homozygous for the other two missense variants already reported among CYP2B6 alleles (p.Lys262Arg and p. Gln172His). Another four missense variants not reported among PharmVar alleles were identified (Table 2).
Normal metabolizers were the most frequent phenotypes in CYP2C19 (n = 205, 45.35%), CYP2C9 (n = 318, 70.51%), and CYP3A4 (n = 422, 94.62%) genes (Figure 2B). In the CYP2C19 gene, similar prevalences of rapid (n = 108, 23.89%) and intermediate (n = 106, 23.45%) metabolizers were observed, and *1 (normal function, n = 573, 63.38%), *17 (increased function, n = 160, 17.70%) and *2 (no function, n = 115, 12.72%) alleles were the most prevalent. We identified eleven missense variants not included among CYP2C19 PharmVar-reported alleles (Table 2). Interestingly, the unreported CYP2C19 p. Thr130Met variant (rs150152656) and the CYP2C19 p. Ile331Val variant (rs3758581) were in the same haplotype, since the individual was homozygous for CYP2C19 p. Ile331Val. For the CYP2C9 and CYP3A4, the *1 allele was the most common among the 10 and 8 different alleles identified, respectively (Supplementary Table S3). Among PharmVar unreported alleles, we found four missense variants in the CYP2C9 gene, with an allele frequency of 0.11%, of which two were found in the same individual (CYP2C9 p. Ile389Thr and p. His410Arg) (Table 2). For the CYP3A4 gene, three missense variants (Table 2) and one frameshift variant (rs1263184572, p. Leu51PhefsTer39) were identified. The frameshift variant had an allele frequency of 0.11% and lacks in silico predictions in SIFT and PolyPhen databases.
On the other hand, poor (n = 270, 59.87%) and intermediate (n = 153, 33.92%) metabolizers were the most frequent phenotypes identified in the CYP3A5 gene (Figure 2B; Supplementary Table S4). In this case, the nonfunctional CYP3A5**3 allele had a prevalence of 70.95% (n = 640). Among the alleles not reported in PharmVar, we identified three missense variants (Table 2) and two stop-gained variants in this gene. The CYP3A5 p. Gln460 variant (rs149664815) was observed in one individual (0.11%), while the CYP3A5 p. Arg268* variant (rs148176345) was detected in three individuals (0.33%).
Finally, in the CYP4F2 gene, the *1/*1, *1/*4, and *1/*17 diplotypes were most prevalent, with no recommendations or predicted phenotypes determined by our analysis (Supplementary Table S3). Moreover, among missense variants in the CYP4F2 gene, the CYP4F2 p. Ala483Gly variant (rs3952537) showed an allele frequency of 4.95% (Table 2). Based on inferred phased genomic data, the CYP4F2 p. Ala483Gly forms a haplotype with CYP4F2 p. Thr472Ala, a variant of the reported *CYP4F2**17 allele.
Across the analyzed pharmacogenes, most individuals were classified as normal metabolizers (Figure 2C). For NUDT15, 92.92% of participants (n = 420) showed a normal phenotype, predominantly the *1/*1 diplotype, while nonfunctional (*2, *3, *9) and uncertain function (*4, *6) alleles were rare. Similarly, for TPMT, the *1/*1 diplotype was the most frequent (n = 401, 88.72%), although 37 individuals carried nonfunctional alleles (*2, *3A, *3C, *8, *9, *24, *34) and were classified as intermediate, possible intermediate, or poor metabolizers. For UGT1A1, the distribution was more balanced, with comparable frequencies of normal (42.0%) and intermediate (41.8%) metabolizers, while decreased-function alleles (*6, *28, *80+*28, *80 + 37) accounted for approximately 37% of the cohort.
In DPYD, intermediate metabolizers were uncommon (n = 9, 2.0%). The nonfunctional *DPYD**2A, decreased function rs67376798-A, and HapB3 variants were each detected in 0.33% of participants, while *7 and rs115232898 were each in 0.11%. In RYR1, malignant hyperthermia susceptibility was identified in only 2 individuals (0.44%), while 98.76% (n = 446) exhibited normal function. Variants of uncertain function (c.652G>A; c.9635A>G; c.12553G>A; c.1598G>A) were identified in 4 participants (0.88%). Moreover, 2 undocumented genetic variations were detected in our sample. Forty-five individuals had a T-allele at the RYR1 c.7089 position, and 1 individual had a T-allele at the RYR1 c.6612 position (rs141646642). Importantly, the RYR1 c.7089C>G and c.6612C>G variants have been described as associated with malignant hyperthermia susceptibility. Detailed allele frequencies are provided in Supplementary Tables S3, S4.
Discussion
4
In this study, we conducted a comprehensive PGx analysis in a cohort of Brazilian patients with MPC and HER2-positive BC using exome sequencing and the PharmCAT. Nearly all participants carried at least one actionable PGx phenotype, underscoring the potential clinical utility of PGx testing in this population. In settings characterized by polypharmacy, narrow therapeutic indices, and cumulative toxicities, preemptive genotyping offers a realistic path to fewer adverse events, more predictable exposures, and more efficient use of supportive care. Evidence from population-scale studies has shown similar clinical relevance across diverse cohorts (Bousman et al., 2025; Hodel et al., 2024; McInnes et al., 2021; Moore et al., 2025), reinforcing the generalizability of our conclusions to routine cancer care. The high prevalence of actionable variants across multiple genes highlights the importance of incorporating PGx testing into treatment decision-making, particularly in oncology, where polypharmacy and drug toxicity are common. Our results revealed substantial variability in the frequency of actionable alleles across pharmacogenes, consistent with findings from a previous study in a Southeastern Brazilian cohort (Bertholim-Nasciben et al., 2023).
ABCG2, encodes the BC resistance protein (BCRP), a major efflux transporter involved in drug predisposition. The common missense variant rs2231142 (Q141K) leads to the substitution of glutamine (Q) by lysine (K) at position 141, resulting in decreased transport activity (Suominen et al., 2023). In our cohort, 17% of individuals carried the T allele associated with decreased function, which may increase exposure to multiple oncology and supportive-care drugs. The clinical consequences are setting- and drug-dependent, and may include high rates of cytopenias, diarrhea, and elevated liver function tests in susceptible patients, in addition to an increased risk of rosuvastatin-induced myopathy, particularly in the presence of other risk factors or drug–drug interactions (Suominen et al., 2023).
The IFNL3/4 rs12979860 T allele is a well-established predictor of response to interferon-based therapy in chronic hepatitis C virus (HCV) infection. Its clinical relevance in oncology, however, remains limited and has not been definitively established. Most evidence derives from studies in patients with chronic HCV infection, where the T allele has been associated with a higher risk of hepatocellular carcinoma and with differences in immune–inflammatory pathways that may influence cancer susceptibility or progression. Beyond this context, its impact on cancer treatment outcomes or toxicity remains exploratory, with no actionable oncology-specific guidelines currently available. Therefore, to date, there is no evidence supporting its role in systemic therapy selection for BC or prostate cancer (PC), and it should not guide oncology decisions (Hou et al., 2019). Conversely, the VKORC1 rs9923231 T-allele, identified in one-third of our cohort, is associated with increased warfarin sensitivity, conferring lower dose requirements. This finding may impact patients with cancer, who often require warfarin for venous thromboembolism prophylaxis or treatment (Johnson et al., 2017).
SLCO1B1 encodes the hepatic uptake transporter Organic Anion Transporting Polypeptide 1B1 (OATP1B1), which mediates the hepatic uptake of several statins. Decreased/poor function (often driven by *5/*15 haplotypes) increases systemic statin exposure and the risk of myopathy, especially with simvastatin and, to a lesser extent, atorvastatin (Ramsey et al., 2014). In our sample, SLCO1B1 exhibited marked allelic heterogeneity, with decreased-function variants accounting for 21.1% of predicted phenotypes, consistent with frequencies reported in Latin American populations (Cooper-DeHoff et al., 2022; Ramsey et al., 2014). OATP1B1 also contributes to the hepatic uptake of docetaxel, and some studies have related SLCO1B1 variants/haplotypes to altered docetaxel pharmacokinetics and toxicity. However, these findings are inconsistent and currently do not support genotype-based dosing (Hjorth et al., 2022; Schmidt et al., 2025).
CYP genes accounted for most of the predicted metabolic variability. For instance, CYP2B6 contributes to the activation and clearance of several drugs commonly used in oncology, such as cyclophosphamide in combination regimens, bupropion, and methadone (Zanger and Klein, 2013). In patients with intermediate or reduced CYP2B6 activity receiving cyclophosphamide, which is frequently used in the neo/adjuvant treatment of BC, closer monitoring for myelosuppression and nausea/vomiting is recommended, along with the evaluation of concomitant medications that may inhibit or induce CYP2B6 activity (Helsby et al., 2021). In our cohort, CYP2B6 exhibited substantial allelic heterogeneity, with 19 different star alleles identified. The prevalence of the *6 allele is consistent with frequencies reported in European and Latin American populations, ranging from 15% to 30% (Langmia et al., 2021; Zanger and Klein, 2013; Zhou and Lauschke, 2022).
The distribution of CYP2C19 alleles in our sample and the resulting phenotype diversity paralleled findings from admixed and European cohorts, reinforcing the genetic heterogeneity of the Brazilian population (Zhou and Lauschke, 2022). CYP2C19 loss-of-function alleles (e.g., *2) reduce clopidogrel activation and increase the risk of stent thrombosis; whereas the *17 allele is associated with increased function, which may increase bleeding risk and reduce proton-pump inhibitor exposure, potentially compromising reflux prophylaxis or ulcer prevention. CYP2C19 activity also influences the metabolism of several antidepressants prescribed for hot flashes, anxiety, or depression (e.g., citalopram/escitalopram, sertraline) (Scott et al., 2013). In the context of MPC treatment, enzalutamide acts as a moderate CYP2C19 inducer, which may further reduce exposure to proton-pump inhibitors and antidepressants in *17 carriers who are already predisposed to faster CYP2C19 metabolism (Gibbons et al., 2015).
CYP3A5 showed marked variability, which aligns with previous findings that CYP3A5 loss-of-function alleles are common in European-derived populations, but less frequent in African populations, where functional alleles are more common (PharmGKB, 2025). *CYP3A5**6 and *7 alleles were slightly more prevalent in our cohort that in European ancestry populations, aligning with previous estimates from Latino populations (Pratt et al., 2023). Although CYP3A4 is the dominant enzyme in the taxane pathway, CYP3A5 loss-of-function alleles may reduce overall CYP3A capacity and exacerbate toxicity when CYP3A4 activity is concurrently inhibited. Moreover, studies have supported an association of PC risk with CYP3A5 variants, as well as with increased CYP3A5 expression (Van Eijk et al., 2019).
For CYP2C9, the prevalence of decreased-function alleles translated into 29.5% of individuals being predicted as poor or intermediate metabolizers, in line with global estimates (Zhou et al., 2023). Although less common in our cohort, CYP2C9 loss-of-function alleles (*3, *6, *33) are known to reduce warfarin clearance, thereby increasing bleeding risk at standard doses (Sanderson et al., 2005).
In genes such as TPMT, NUDT15, and DPYD, although the prevalence of nonfunctional alleles was low, the clinical implications remain substantial, as carriers are at a significantly increased risk of severe toxicity when exposed to thiopurines or fluoropyrimidines. Particularly, the NUDT15 nonfunctional alleles *2, *3, and *9 —associated with an intermediate metabolizer phenotype—were rare, and the data are consistent with findings in European, Amazonian Amerindian, and admixed Brazilian cohorts (Karczewski et al., 2020; Naslavsky et al., 2022; Rodrigues et al., 2020). Similarly, TPMT and DPYD poor and intermediate metabolizers were less frequent, confirming previous findings in Brazilian cohorts (Bertholim-Nasciben et al., 2023; Naslavsky et al., 2022). There is robust evidence that genotype-guided fluoropyrimidine dosing significantly reduces grade ≥3 toxicity (Amstutz et al., 2018) and converging regulatory frameworks support pre-treatment DPYD testing (Keen et al., 2025). Routine implementation of this practice is warranted in Brazilian BC care settings, with dose reductions according to international guidelines (Amstutz et al., 2018). Finally, the UGT1A1 gene showed decreased-function alleles in approximately 37% of participants, resulting in a large proportion of predicted intermediate metabolizers, consistent with a previous study in a Brazilian population (Bertholim-Nasciben et al., 2023). This may be clinically relevant when using drugs whose exposure/toxicity is driven by UGT1A1 activity, such as sacituzumab govitecan and irinotecan (Karas and Innocenti, 2022).
The high prevalence of actionable PGx profiles in our sample (99.3%) has significant implications for precision oncology within the Brazilian public health system (SUS). Patients with BC and PC are frequently exposed to drugs with narrow therapeutic indices and high risk of adverse reactions, including tamoxifen, fluoropyrimidines, taxanes, statins, and anticoagulants (Al-Mahayri et al., 2020; Basak et al., 2021). In this context, DPYD, NUDT15 and TPMT variants may predict chemotherapy-related toxicity (Larrue et al., 2024; Yang et al., 2015). Since 2020, the European Medicines Agency has recommended dihydropyrimidine dehydrogenase (DPD) deficiency testing before the use of fluorouracil and capecitabine (De With et al., 2023), and the U.S. Food and Drug Administration has recently updated the product labeling of these drugs, to increased awareness of DPD deficiency (FDA, 2025). Additionally, testing for TPMT and NUDT15 genes has been funded by the United Kingdom National Health Service for patients with acute lymphoblastic leukemia treated with purine analogs (NHS, 2025). In Brazil, there is no formal recommendation or mandatory coverage for TPMT, NUDT15, or DPYD testing by national health authorities.
The relevance of our results lies in the admixed genetic background of the Brazilian population, characterized by different contributions from European, African, and Native American ancestries. Our ancestry analysis confirmed this admixture and identified rare and previously unreported variants, further supporting the notion that admixed populations remain underrepresented in global PGx databases (Mychaleckyj et al., 2017; Secolin et al., 2019). This underrepresentation may limit the accuracy of phenotype prediction tools and clinical guidelines, which have been developed using predominantly European reference populations. Our study, therefore, contributes to filling this knowledge gap by providing large-scale data from a representative Brazilian cohort. From a clinical perspective, several of the PGx variants identified in this study are included in existing pharmacogenomic guidelines and may help inform treatment decisions. Although formal clinical impact was not evaluated here, these findings highlight the potential relevance of integrating PGx information into public healthcare settings.
Strengths of our study include the use of exome sequencing, which enabled the detection of both common and rare alleles, the multicenter recruitment strategy ensuring representation from all Brazilian macro-regions, and the application of standardized tools (PharmCAT and CPIC® guidelines) for variant interpretation. Nevertheless, some limitations must be acknowledged. First, CYP2D6, a clinically relevant pharmacogene, was not assessed due to technical challenges in accurately detecting copy number and structural variants using exome sequencing. More broadly, exome sequencing primarily targets coding regions and does not reliably capture promoter, intronic, or other structural variants, which may also be functionally relevant in pharmacogenomics. As a result, regulatory and structural variation contributing to drug response may not be fully represented in this study. Second, although novel missense variants were identified, their functional impact could not be reliably inferred based solely on sequence data. Functional validation and predictive modeling will be required to determine their potential clinical relevance. Third, the study population consisted exclusively of patients with cancer, which may not fully represent the general Brazilian population. Finally, the predominance of European ancestry in our cohort, while reflective of certain regions of Brazil, may not capture the full extent of variability across the country.
In conclusion, our study highlights PGx variability in a large admixed Brazilian cohort, consistent with global data but shaped by the unique ancestry composition of the population. Our data also demonstrate that nearly all individuals harbor actionable PGx phenotypes. Incorporating PGx testing into SUS protocols could improve patient safety, reduce adverse drug reactions and hospitalizations, and optimize therapeutic outcomes. However, such implementation requires overcoming barriers such as financial constraints, infrastructure, and training healthcare professionals. These results also emphasize the urgent need to continue generating genomic data from underrepresented populations. Such efforts will be essential for the equitable advancement of precision medicine worldwide.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Al-Mahayri Z. N. Patrinos G. P. Ali B. R. (2020). Toxicity and pharmacogenomic biomarkers in breast cancer chemotherapy. Front. Pharmacol. 11, 445. 10.3389/fphar.2020.00445 32351390 PMC 7174767 · doi ↗ · pubmed ↗
- 2Alexander D. H. Novembre J. Lange K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. 10.1101/gr.094052.109 19648217 PMC 2752134 · doi ↗ · pubmed ↗
- 3Amstutz U. Henricks L. M. Offer S. M. Barbarino J. Schellens J. H. M. Swen J. J. (2018). Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther. 103, 210–216. 10.1002/cpt.911 29152729 PMC 5760397 · doi ↗ · pubmed ↗
- 4Basak D. Arrighi S. Darwiche Y. Deb S. (2021). Comparison of anticancer drug toxicities: Paradigm shift in adverse effect profile. Life 12, 48. 10.3390/life 12010048 35054441 PMC 8777973 · doi ↗ · pubmed ↗
- 5Bergström A. Mc Carthy S. A. Hui R. Almarri M. A. Ayub Q. Danecek P. (2020). Insights into human genetic variation and population history from 929 diverse genomes. Science 367, eaay 5012. 10.1126/science.aay 5012 32193295 PMC 7115999 · doi ↗ · pubmed ↗
- 6Bertholim-Nasciben L. Scliar M. O. Debortoli G. Thiruvahindrapuram B. Scherer S. W. Duarte Y. A. O. (2023). Characterization of pharmacogenomic variants in a Brazilian admixed cohort of elderly individuals based on whole-genome sequencing data. Front. Pharmacol. 14, 1178715. 10.3389/fphar.2023.1178715 37234706 PMC 10206227 · doi ↗ · pubmed ↗
- 7Bousman C. A. Narang A. Al Bkhetan Z. Woods R. L. Orchard S. G. Owen A. J. (2025). Prevalence of actionable pharmacogenetic genotype frequencies, cautionary medication use, and polypharmacy in community‐dwelling older adults. Clin. Pharmacol. Ther. 118, 337–342. 10.1002/cpt.3702 40304392 PMC 12272320 · doi ↗ · pubmed ↗
- 8Bush W. Crosslin D. Owusu‐Obeng A. Wallace J. Almoguera B. Basford M. (2016). Genetic variation among 82 pharmacogenes: the PGR Nseq data from the e MERGE network. Clin. Pharmacol. Ther. 100, 160–169. 10.1002/cpt.350 26857349 PMC 5010878 · doi ↗ · pubmed ↗