Disrupting PDGFRA-driven immune evasion in glioma: vaccine-based strategies on the horizon
Xialin Zhang, Xinwei Li, Ran Cui, Xinlin Yu, Zihan Zhang, Zhongxiang Luo, Gang Chen, Sheng Lin

TL;DR
This paper explores how targeting PDGFRA in gliomas could help overcome immune evasion and improve treatment outcomes through vaccine-based strategies.
Contribution
The paper introduces PDGFRA-targeted vaccines as a novel approach to simultaneously suppress tumor growth and reverse immune evasion in gliomas.
Findings
PDGFRA signaling promotes immunosuppression in gliomas through vascular abnormalities and metabolic reprogramming.
Vaccines targeting PDGFRA may reverse immune evasion while suppressing tumor proliferation.
Future strategies include multi-omics, nanodelivery systems, and combinations with checkpoint inhibitors to enhance efficacy.
Abstract
Gliomas, the most aggressive primary brain tumors, present a formidable challenge in neuro-oncology, characterized by infiltrative growth, high recurrence rates, and a profoundly immunosuppressive microenvironment that severely limits the efficacy of current treatments. Platelet-derived growth factor receptor alpha (PDGFRA) has emerged as a pivotal oncogenic driver in gliomas, not only promoting cellular proliferation and angiogenesis but critically orchestrating complex immune evasion mechanisms. Understanding how PDGFRA shapes this immunosuppressive landscape is paramount for developing effective immunotherapies, especially given the minimal response rates of gliomas to conventional checkpoint inhibitors. PDGFRA signaling actively remodels the glioma microenvironment, contributing to vascular abnormalities (e.g., via the PDGFRA-Endocan-MYC axis), metabolic reprogramming that impairs T…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
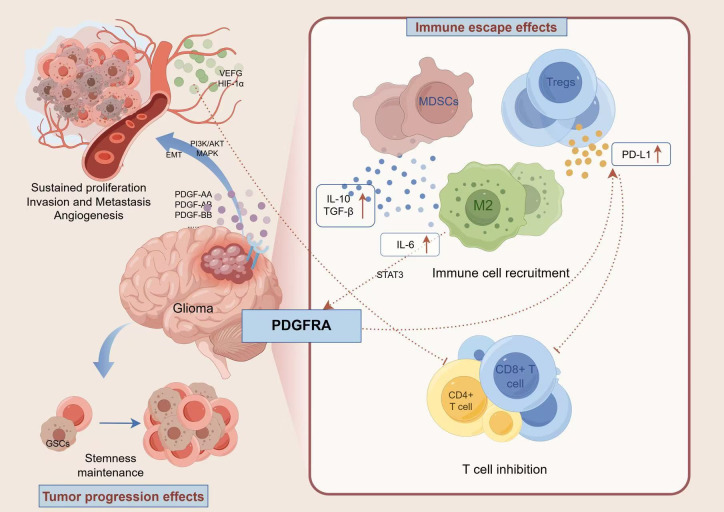 Figure 1
Figure 1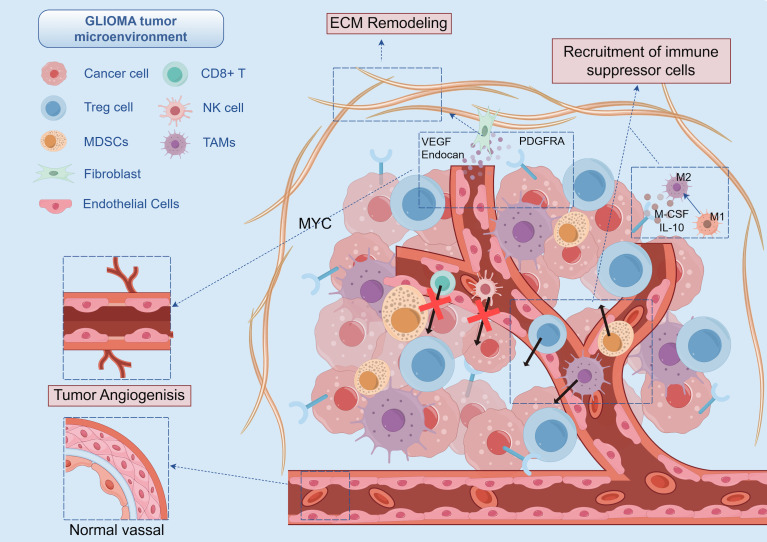 Figure 2
Figure 2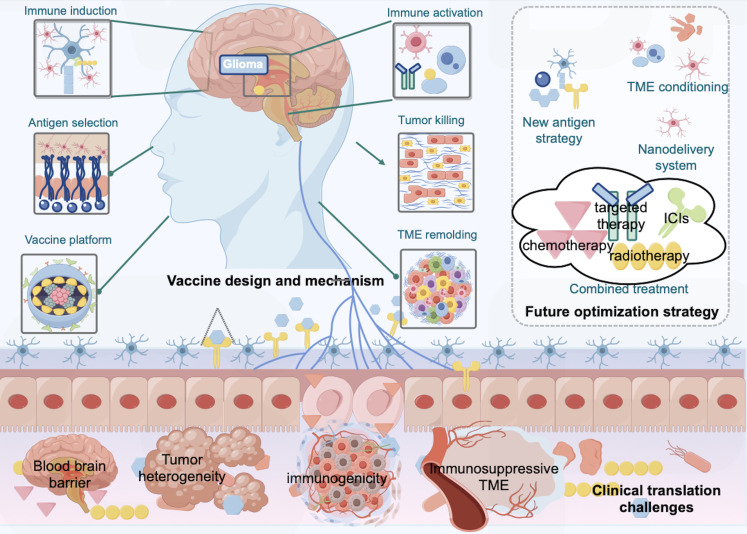 Figure 3
Figure 3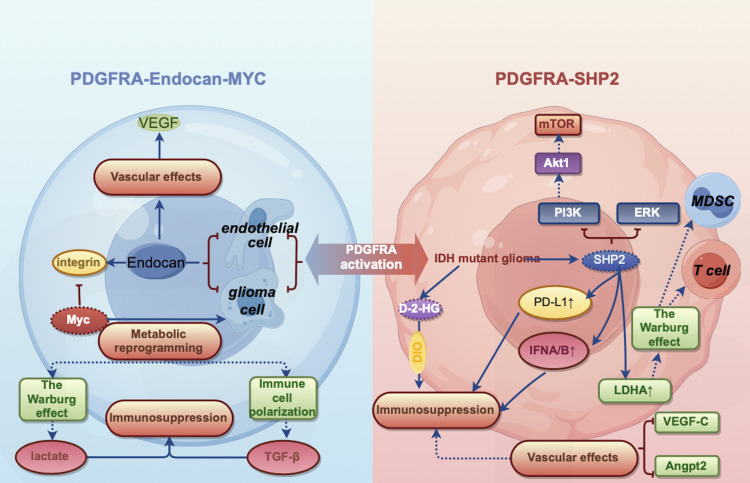 Figure 4
Figure 4| Model name | Mouse strain | Vaccine format | Outcomes | Reference |
|---|---|---|---|---|
| Syngeneic mouse models | C57BL/6 mice | peptide | Increased survival rate | ( |
| Syngeneic mouse models | C57BL/6J mice | peptide | Increased survival rate | ( |
| Syngeneic mouse models | C57BL/6N mice | peptide | Increased survival rate | ( |
| Humanized mouse models | A2.DR1 mice | peptide | Vaccination reduces growth of tumors | ( |
| Syngeneic mouse models | C57BL/6 mice | peptide | Increased survival rate | ( |
| Syngeneic mouse models | C57BL/6 mice | mRNA | mOS:40 vs 31 days | ( |
| Spontaneous glioma | Dog | mRNA | mOS:139 vs 35 days | ( |
| Humanized mouse models | NOG mice | Dendritic cell | mOS:60 vs 40 days | ( |
| Syngeneic mouse models | C57BL/6 mice | Dendritic cell | Increased survival rate | ( |
| Syngeneic mouse models | C57BL/6 mice | Dendritic cell | mOS:38.5 vs 26.8 days | ( |
| Trial | Phases | Vaccine | Vaccine format | Status | Enrollment | Key outcomes | Reference |
|---|---|---|---|---|---|---|---|
| I | TetGAA peptide vaccine | Peptide | Completed | 60 | mPFS:9.9 months | ( | |
| I | IMA950 | Peptide | Completed | 45 | PFS-6:74% | ( | |
| I | SurVaxM | Peptide | Completed | 9 | mPFS:17.6 weeks mOS:86.6 weeks | ( | |
| I | IDH1-vac | Peptide | Completed | 39 | ORR:84.4% | ( | |
| I | TAA-based tumor vaccines | Dendritic cell | Completed | 10 | mOS:19 months | ( | |
| I | APVAC | DNA/RNA | Completed | 16 | mPFS:14.2 months | ( | |
| I/II | HSPPC-96 | Peptide | Completed | 41 | mOS:42.6 weeks | ( | |
| I/II | WT1-mRNA/DC | Dendritic cell | Completed | 48 | mOS:43.7 months | ( | |
| II | AV-GBM-1 | Dendritic cell | Unknown | 55 | mPFS:10.4 months | ( | |
| III | DCVax-L | Dendritic cell | Unknown | 331 | nGBM:mOS 19.3 months | ( | |
| II | SurVaxM | Peptide | Active | 66 | mPFS:11.4 months | ( |
| Antigen source | Specific peptide/Variation | HLA restriction | Discovery method | Evidence |
|---|---|---|---|---|
| Wild-type PDGFRA | IMA950 (Multi-peptide cocktail) | HLA-A*02 | Ligandome analysis (Mass Spec) | Included in the IMA950 vaccine; naturally presented on GBM surface in patients with PDGFRA overexpression (Schoor et al., Cancer Res). |
| Wild-type PDGFRA | KTS (and other APVAC1 peptides) | Class I (A02, A24) | GAPVAC Trial (Immunopeptidomics) | Personalized vaccine targets identified directly from patient tumor tissue; PDGFRA peptides were among the frequent “warehouse” targets selected (Hilf et al., Nature 2019). |
| Mutant PDGFRA | D842V (Exon 18) | HLA-A*02 (Predicted) | In silico NetMHC prediction | The most common activating mutation in GBM; generates a high-affinity neoepitope distinct from wild-type, minimizing autoimmunity risk (Pre-clinical rationale). |
| Fusion/Del | PDGFRA-Δ8/9 (Deletion) | Patient-specific | Transcriptome Sequencing | In-frame deletions (e.g., Gas7-PDGFRA or exon deletions) create novel junctional epitopes suitable for personalized mRNA or peptide vaccines (Ozawa et al., Genes Dev). |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlioma Diagnosis and Treatment · Cancer Immunotherapy and Biomarkers · Immune cells in cancer
Introduction
Gliomas represent a formidable challenge in neuro-oncology, with poor survival outcomes despite multimodal treatment approaches (1). Their therapeutic resistance stems from infiltrative growth patterns and sophisticated immune evasion mechanisms that create a profoundly “cold” tumor microenvironment (2, 3). Platelet-derived growth factor receptor alpha (PDGFRA) has emerged as a critical oncogenic driver in gliomas (4). Beyond promoting cellular proliferation and angiogenesis, PDGFRA orchestrates complex immune evasion mechanisms, positioning it at the intersection of oncogenic signaling and immunosuppression (5). Consequently, understanding and disrupting PDGFRA-mediated immune evasion holds significant promise for the development of novel, translational immunotherapeutic interventions for this challenging disease.
Immunotherapy has transformed cancer treatment paradigms, yet its efficacy in gliomas has been markedly limited, with checkpoint inhibitor response rates generally remaining below 10% (6). This resistance stems from a complex interplay of factors, including the intrinsic impermeability of the blood-brain barrier (BBB), a generally low tumor mutational burden, and a profoundly immunosuppressive tumor microenvironment. This microenvironment is characterized by the abundance of immunosuppressive cells, such as regulatory T-cells and myeloid-derived suppressor cells, alongside M2-polarized tumor-associated macrophages (7). Crucially, PDGFRA activation plays a pivotal role in shaping this adverse immune landscape. Through intricate mechanisms, PDGFRA signaling contributes to the modulation of cytokine networks, orchestrates metabolic reprogramming that impairs T-cell effector functions, and promotes vascular remodeling that restricts essential immune cell infiltration into the tumor core (8, 9). Consequently, targeting PDGFRA presents a compelling therapeutic opportunity, as it may simultaneously inhibit oncogenic signaling driving tumor progression while actively reversing the tumor-mediated immunosuppression that thwarts effective anti-tumor immunity (10, 11).
This review examines PDGFRA’s multifaceted role in glioma pathophysiology, emphasizing its immunomodulatory functions. We delineate PDGFRA signaling networks and explore how it orchestrates immune evasion through three critical mechanisms: vascular remodeling, immune cell polarization, and metabolic reprogramming. We assess current PDGFRA-targeted interventions, including small molecule inhibitors and emerging vaccine-based approaches, critically evaluate translational challenges, and outline promising directions in personalized immunotherapy. Through this analysis, we provide a framework for developing PDGFRA-targeted interventions that may overcome the formidable challenge of immune evasion in gliomas.
Role of PDGFRA signaling pathway in gliomas
Structure, function, and dysregulation of PDGFRA in gliomas
PDGFRA is a transmembrane receptor tyrosine kinase (RTK) and a member of the type III RTK family, playing a critical role in biological processes, particularly during development. Physiologically, PDGFRA is crucial for neural development, governing the proliferation and differentiation of neural progenitor cells, with a notable essential role in the development of oligodendrocyte progenitor cells (OPCs) (12). In the adult brain, PDGFRA expression significantly diminishes, being primarily confined to a subset of progenitor cells and perivascular cells where it contributes to neuroglia homeostasis and the maintenance of BBB integrity (13). However, during pathological conditions, including neuronal injury and glioma, PDGFRA expression is substantially upregulated, initiating downstream signaling cascades. This upregulation exhibits pronounced spatiotemporal specificity, with a particular prevalence observed in neuroglial progenitor cells and astrocytes (14). Aberrant activation of PDGFRA is a hallmark of many gliomas, driven by mechanisms such as gene amplification or specific mutations (e.g., D842V, V561D) that lead to conformational changes in the kinase domain, resulting in ligand-independent, persistent activation (5).
PDGFRA signaling activation primarily occurs through ligand-dependent and ligand-independent pathways. In ligand-dependent activation, PDGF ligand binding induces receptor dimerization, precipitating autophosphorylation of the intracellular kinase domain. The phosphorylated receptor subsequently recruits diverse downstream signaling molecules (5, 15). For instance, within hypoxic microenvironments, PDGF ligand expression and secretion are regulated by the transcription factor HIF-1α; concurrently, the downstream signaling molecule PTEN negatively regulates the PI3K/AKT pathway (5). Ligand-independent activation mechanisms have also been identified in gliomas. These include epigenetic modifications such as altered histone modifications and DNA methylation influencing PDGFRA gene expression (16). Furthermore, diminished miR-34a expression leads to enhanced PDGFRA mRNA stability (17) and cytokine signaling, such as IL-6 secreted by tumor-associated macrophages (TAMs), can augment PDGFRA expression via STAT3-mediated positive feedback loops (18). The dysregulation of these regulatory mechanisms in gliomas results in the aberrant activation of the PDGFRA signaling pathway. It is important to note that co-activation of PDGFRA and EGFR can trigger compensatory resistance in signaling pathways, potentially limiting the efficacy of therapies targeting only a single pathway (19).
Following activation, PDGFRA principally transduces signals through three critical signaling cascades: initially, the PI3K/AKT/mTOR pathway drives cellular survival and metabolic reprogramming; subsequently, the RAS/RAF/MEK/ERK signaling cascade propels cellular proliferation and differentiation processes; finally, the JAK/STAT pathway mediates the regulation of gene transcription and inflammatory responses (20–22). Additionally, recent discoveries indicate that PDGFRA can participate in tumor immune evasion mechanisms via non-canonical pathways, such as direct interaction with PD-L1 (23). The regulation of these signaling pathways encompasses multi-tiered mechanisms, including dephosphorylation mediated by protein tyrosine phosphatases (PTPs), receptor internalization and degradation, miRNA-regulated expression, and crosstalk with other RTKs (e.g., EGFR) (24).
PDGFRA genetic alterations manifest in diverse forms, predominantly comprising gene amplification (approximately 15%), activating mutations (approximately 5-7%), and rare KIT-PDGFRA fusions (25). The fifth edition of the WHO Classification of Central Nervous System tumors, published in 2021, incorporates molecular characteristics as essential elements in glioma classification, with PDGFRA genetic alterations constituting a pivotal feature in molecular subtyping. PDGFRA alterations exhibit heterogeneous distribution across glioma subtypes. Within glioblastomas, approximately 26% of IDH-wildtype cases and 11% of IDH-mutant cases demonstrate PDGFRA overexpression or genetic alterations (26). Single-cell sequencing and spatial transcriptomic analyses have revealed PDGFRA alterations predominantly enriched in “proneural” and “mesenchymal” molecular subtypes, which typically manifest heightened malignancy and inferior prognostic outcomes (27). PDGFRA exhibits complex interactions with classical glioma genetic markers, displaying significant co-occurrence or mutual exclusivity relationships with IDH1/2 mutations, 1p/19q codeletion, and MGMT promoter methylation. Notably, PDGFRA amplification occurs more frequently in IDH-wildtype glioblastomas and typically demonstrates mutual exclusivity with EGFR amplification (28). PDGFRA mutations (e.g., D842V, R841K) are detected in approximately 5-7% of gliomas, predominantly localized to the intracellular kinase domain, resulting in constitutive receptor activation (29). Additionally, 4q12 amplifications involving PDGFRA, KIT, and KDR genes have been identified in a subset of gliomas. Significantly, a distinct subgroup of pediatric high-grade gliomas—diffuse intrinsic pontine glioma (DIPG)—frequently harbors PDGFRA mutations or fusions, co-occurring with epigenetic abnormalities such as H3K27M, indicating complex molecular pathogenetic mechanisms (30). Multi-omic analyses based on the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases demonstrate that PDGFRA-altered gliomas exhibit distinctive metabolic reprogramming characteristics (enhanced glutamine dependency), potentially serving as actionable therapeutic targets and providing rationale for personalized treatment approaches (31, 32) (Figure 1).
PDGFRA signaling: A dual orchestrator of glioma progression and immune evasion. PDGFRA is aberrantly activated in gliomas due to gene amplification, mutations (such as D842V), or extracellular domain deletion mutations. It drives cell proliferation, metabolic reprogramming, and EMT-like invasion by activating downstream signaling pathways including PI3K/AKT and RAS/RAF/MEK. Meanwhile, PDGFRA upregulates PD-L1, recruits immunosuppressive cells (e.g., regulatory T cells), and downregulates MHC-I expression to establish an immunosuppressive microenvironment. It also mediates immune evasion through direct interaction with PD-L1. Genetic variations of PDGFRA (amplification, activating mutations, fusions) are of critical significance in molecular subtyping of gliomas, particularly enriched in highly malignant subtypes.
PDGFRA-mediated immune evasion
The PDGFRA signaling pathway is a critical regulator in glioma pathogenesis, implicated in tumor initiation, progression, and importantly, in orchestrating immune evasion. Upregulation of PDGFRA is observed across various aggressive glioma subtypes, including diffuse midline gliomas and pediatric high-grade gliomas, acting as a key driver in the transition from low-grade to high-grade disease (33). Notably, extracellular domain deletion mutations in PDGFRA enhance its synergistic interaction with immunosuppressive cytokines, such as TGF-β,within the tumor microenvironment, thereby facilitating immune evasion (30). During tumor initiation, PDGFRA promotes cellular proliferation and survival, partly by activating the PI3K/AKT and MAPK pathways, which upregulate anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL) and enhance cell cycle progression (34). Single-cell sequencing data reveal a strong correlation between high PDGFRA expression in glioma cells and T cell exhaustion phenotypes (35).
PDGFRA signaling recruits immunosuppressive cells, including regulatory T cells (Tregs), TAMs, and myeloid-derived suppressor cells (MDSCs). These recruited cells, in turn, secrete immunosuppressive cytokines such as IL-10 and TGF-β. PDGFRA can also downregulate MHC-I expression on tumor cells, collectively creating an immunosuppressive milieu that blunts anti-tumor immune responses (36). This profound impact on the immune landscape provides a strong rationale for targeting PDGFRA in the development of effective glioma vaccine strategies.
PDGFRA-mediated tumor immune microenvironment remodeling
PDGFRA’s core role in regulating the immune microenvironment
PDGFRA critically orchestrates tumor-immune cell interactions through multiple signaling pathways, significantly shaping the tumor immune microenvironment (TME). Within neoplastic cells, PDGFRA signaling upregulates chemokines like CCL2 and CCL5, which promote monocyte migration and M2-like polarization (37). Transcriptomic analysis of glioblastoma (GBM) with PDGFRA alterations reveals enrichment in immune evasion pathways, including the downregulation of Major Histocompatibility Complex (MHC) class I molecules and the upregulation of immune checkpoint ligands such as PD-L1 (38). Crucially, PDGFRA plays a multifaceted role in fostering an immunosuppressive tumor microenvironment (TME) that hinders therapeutic efficacy, including immune-based interventions. PDGFRA overexpression has been linked to increased PD-L1 expression on glioma cells, leading to suppressed CD8+ T cell activity. It also enhances the self-renewal and differentiation capacity of glioma stem cells (GSCs), contributing to chemo- and radioresistance (39, 40).
Furthermore, in patient-derived GBM models, the PDGFA-PDGFRA signaling axis elevates expression of the tryptophan-degrading enzyme indoleamine 2,3-dioxygenase 1 (IDO1). This enzymatic activity depletes tryptophan, a crucial nutrient for T cell function, thereby suppressing T cell activity (41). Concurrently, PDGFRA-mediated angiogenesis contributes to dysregulated vascular architecture, impeding immune cell infiltration. For instance, in models where Endocan, a proteoglycan secreted by endothelial cells, activates PDGFRA, it enhances vascular permeability and fosters hypoxic microenvironments (42). These hypoxic regions further drive HIF-1α-dependent secretion of VEGF and TGF-β, thereby intensifying immunosuppression (43).
Clinical data from TCGA demonstrates that GBM exhibiting high PDGFRA expression manifest reduced CD8+ T cell infiltration, elevated expression of immunosuppressive cytokines, and correlate with inferior survival outcomes (44). Notably, the co-expression of PDGFRA with the receptor tyrosine kinase EphA2 exacerbates this phenotype by promoting epithelial-mesenchymal transition (EMT) and further attenuating T cell chemotaxis. Collectively, these observations highlight PDGFRA’s central role as a regulator of tumor microenvironment architecture, integrating intrinsic tumor cell signaling with systemic immune dysregulation.
PDGFRA-driven vascular remodeling and the dual roles of blood and lymphatic vessels
PDGFRA-driven vascular remodeling in gliomas creates barriers to immune infiltration. Endocan (Esm1) from endothelial cells activates tumor PDGFRA, triggering MYC-dependent vascular sprouting, basement membrane changes, and leaky vessels with poor pericyte coverage, impairing perfusion and immune extravasation (42, 45). Esm1 knockout reduces permeability and boosts T cell infiltration (46). Genomic analysis reveals that PDGFRA is frequently co-amplified with the vascular regulator KDR (VEGFR2) and KIT at the 4q12 locus in high-grade gliomas, creating a dependency on this oncogenic axis. Targeting this niche with avapritinib, a highly CNS-penetrant PDGFRA/KIT inhibitor, demonstrated significant efficacy in a clinical cohort of pediatric patients (n=8) harboring PDGFRA amplifications or exon 18 mutations, achieving objective radiographic responses in 42% of evaluable cases (45). Mechanistically, PDGFRA inhibition can indirectly reduce VEGF secretion and stabilize vascular structure by inhibiting the PDGFRA signaling axis.
PDGFRA-mediated angiogenesis has bidirectional effects. Pathological angiogenesis forms immunosuppressive barriers, hinders drug delivery, and aids tumor progression/metastasis; vascular normalization improves tumor perfusion, immune cell infiltration, and therapeutic synergy (47, 48). Angiogenesis and lymphangiogenesis collectively exert dual roles. Normalized blood vessels enhance drug delivery efficiency and therapeutic efficacy, while aberrant angiogenesis drives tumor growth and drug resistance. The crosstalk between blood vessels and lymphatics modulates this process, either clearing tumor-derived factors or facilitating tumor metastasis through the PDGFRA/VHL/HIF-VEGF signaling pathway (49, 50). Monotherapy prompts bypass (VEGFR2/FGFR1 upregulation), lymphangiogenic compensation (VEGF-C/PDGF-BB), and immune escape. Vaccines or combination therapies should co-target pathway redundancies or lymphangiogenesis to prevent drug resistance.
Unlike anti-VEGF therapies that exacerbate tumor hypoxia while suppressing angiogenesis, PDGFRA inhibition induces vascular normalization by disrupting MYC signaling, thereby improving tumor perfusion and anti-tumor immunity. PDGFRA vaccines outperform EGFRvIII vaccines due to their broad expression profile, dual effects of oncogenic pathway disruption and immune modulation, and lower escape rate; they show promise despite tumor heterogeneity, though further clinical validation is needed.
PDGFRA signaling: sculpting immune cell polarization and function in the tumor microenvironment
PDGFRA signaling pathways critically influence the differentiation and functional polarization of immune cells within the tumor microenvironment. In myeloid lineage cells, PDGFRA activation promotes differentiation into MDSCs. These MDSCs suppress T cell responses by producing nitric oxide and arginase (51). Notably, in a PDGFRA-driven glioblastoma murine model, eliminating PDGFRA-expressing myeloid cells led to enhanced T cell effector functions and extended survival (52).
Within the tumor microenvironment, high PDGFRA expression in tumors correlates with a prevalent M2-like phenotype in TAMs, characterized by increased CD206 expression and reduced inflammatory markers. This M2 polarization is promoted by PDGFRA-driven secretion of M-CSF and IL-10 from tumor cells. Co-culture experiments have demonstrated that inhibiting PDGFRA can reverse these M2 phenotypic markers (53).
For T cells, the binding of soluble ligands like PDGFA to PDGFRA impairs effector differentiation and drives T cell exhaustion. PDGFA-exposed CD8^+^T cells exhibit reduced IFN-γ production and increased PD-1 expression, a phenotype that can be restored by PDGFRA blockade (54). This impairment of T cell function is particularly severe in tumors with PDGFRA/EPHA2 co-amplification, where combined receptor signaling further exacerbates T cell dysfunction (55) (Figure 2).
PDGFRA-mediated remodeling of glioma microenvironment: Vascular abnormalities and immune cell exclusion. PDGFRA drives angiogenesis sprouting and basement membrane remodeling in gliomas via the Endocan-PDGFRA-MYC axis, forming immature vascular networks. This not only hinders immune cell extravasation but also reduces vascular stability by decreasing pericyte coverage. Meanwhile, PDGFRA activation in pericytes promotes their differentiation into myofibroblasts, which secrete extracellular matrix to form a physical barrier. Additionally, PDGFRA signaling directly promotes myeloid cell differentiation into MDSCs and polarizes TAMs toward an M2-like phenotype. It also impairs T cell effector functions and induces exhaustion through binding to PDGFA ligands.
Regulatory interactions between PDGFRA signaling and the metabolism-immune axis
PDGFRA signaling integrates metabolic reprogramming and immunosuppression
PDGFRA functions as a key receptor tyrosine kinase, integrating cellular metabolism and immune responses. In glioblastoma specimens with high PDGFRA expression, genes associated with glycolysis (e.g., HK2, LDHA) are coordinately upregulated with immune checkpoint molecules (e.g., PD-L1, CTLA-4) (42). Mechanistically, PDGFRA phosphorylation activates AKT, which subsequently phosphorylates mTORC1. This cascade promotes the translation of glucose transporter GLUT1 via S6K1, while simultaneously inhibiting mitochondrial oxidative phosphorylation. This metabolic shift not only enhances tumor cell proliferation but also suppresses T cell function by creating a locally glucose-depleted and lactate-rich environment (56, 57).
PDGFRA signaling additionally modulates tumor microenvironment remodeling via microglia-mediated immune interactions. In IDH-mutant gliomas, the aberrantly activated PDGFRA-SHP2 axis synergizes with RAS-MAPK signaling to drive PD-L1 overexpression. This activation reprograms immune cell phenotypes and dampens therapeutic efficacy, establishing a “signaling-immunosuppression” feedback loop targetable by bionic nanoplatforms (45, 58–60). Studies using patient-derived IDH-mutant glioma stem cells (GSCs) demonstrate that the SHP2 inhibitor SHP099 can reverse PDGFRA-mediated MYC phosphorylation. This intervention restores oxidative phosphorylation, reduces IL-10 secretion, and reactivates cytotoxic function in CD8^+^ T cells (58). Recent studies reveals that PDGFRA^+^ tumor cell subpopulations exhibit co-expression networks between glycolysis gene modules and M2 macrophage polarization markers (such as CD163 and MRC1), suggesting metabolic reprogramming directly orchestrates myeloid cell phenotypes (55).
Regulatory role of the PDGFRA-endocan-MYC axis in the vascular-metabolic microenvironment
Endocan, secreted by tumor vascular endothelial cells, regulates tumor vascularity and spatial phenotype via direct binding and activation of PDGFRA on glioblastoma cells (46). Endocan-PDGFRA signaling activates downstream PI3K and ERK pathways and enhances chromatin accessibility at the Myc promoter, resulting in stable upregulation of MYC expression (61). This signaling axis is essential for establishing the hypervascular phenotype characteristic of GBM and confers radioprotection, consistent with the role of the perivascular niche in therapeutic resistance (62, 63). Distinct from mechanisms promoting immune exclusion (no significant differences in CD8+ T cell infiltration were observed between Esm1 wild-type and knockout tumors), the Endocan-PDGFRA-MYC axis primarily functions to maintain GBM cells in a proliferative, proneural state at the perivascular niche, thereby preventing the stress-induced mesenchymal differentiation typically observed in the tumor core (42, 64, 65).
PDGFRA-mediated tumor-vascular crosstalk represents a critical determinant of glioblastoma progression and therapeutic resistance (66), primarily manifested through its regulation of intratumoral heterogeneity and plasticity rather than direct immune suppression. Investigations demonstrate that the Endocan-PDGFRA axis supports a reciprocal feedback loop where endothelial-secreted factors drive GBM proliferation and promote further neovascularization. Consequently, inhibition of PDGFRA signaling (e.g., with ponatinib) effectively disrupts this vascular-tumor support system, downregulates MYC, and reduces tumor growth specifically in Endocan-rich microenvironments, highlighting the Endocan-PDGFRA axis as a potential therapeutic target to subdue GBM recurrence.
Metabolic-immune reprogramming by the PDGFRA-SHP2 axis in IDH-mutant gliomas
Isocitrate dehydrogenase (IDH) mutations activate PDGFRA via epigenetic mechanisms, driving distinct metabolic-immune interaction patterns (67). IDH mutations result in accumulation of the oncometabolite D-2-hydroxyglutarate (D-2-HG), which competitively inhibits α-ketoglutarate (α-KG)-dependent dioxygenases, thereby disrupting epigenetic regulatory mechanisms and consequent gene expression profiles. These metabolic alterations not only promote tumorigenesis and progression but also enhance immunosuppressive states through modulation of the immune microenvironment.
The PDGFRA-SHP2 axis plays a pivotal role in this process (68). PDGFRA activation, via SHP2, further influences downstream signaling pathways, including ERK and PI3K/AKT/mTOR cascades, which are critical for neoplastic cell proliferation, survival, and metabolic reprogramming. Additionally, the PDGFRA-SHP2 axis promotes the establishment of an immunosuppressive microenvironment through upregulation of immune checkpoint molecules and suppression of genes associated with anti-tumor immune responses (including IFNA/B gene clusters) (58).
Metabolically, the PDGFRA-SHP2 axis enhances lactate dehydrogenase A (LDHA) expression, augmenting lactate production and consequent acidification of the tumor microenvironment (69). This acidic milieu not only suppresses T cell functionality but also promotes accumulation of immunosuppressive cells, notably MDSCs. Concurrently, the PDGFRA-SHP2 axis modulates expression of angiogenic factors, including VEGF-C and Angpt2, facilitating aberrant angiogenesis and further restricting immune cell infiltration (70) (Figure 3).
Crosstalk of PDGFRA signaling with metabolism-immunity axis: Unveiling Endocan-MYC and SHP2 pathways. In gliomas, PDGFRA drives the formation of an immunosuppressive metabolic microenvironment through dual signaling axes: In the PDGFRA-Endocan-MYC axis (left panel), PDGFRA activation promotes Endocan secretion from endothelial/tumor cells, triggering the integrin-MYC signaling cascade that enhances glycolysis (lactate accumulation) and glutaminolysis (kynurenine production), directly suppressing T cell activity and inducing Treg differentiation. In the PDGFRA-SHP2 axis (right panel, IDH-mutant context), PDGFRA activates SHP2 phosphatase, which synergizes with the oncometabolite 2-HG to disrupt mitochondrial metabolism (e.g., TCA cycle dysfunction), leading to CD8+ T cell exhaustion and PD-1 overexpression. These dual axes cooperatively induce metabolic competition imbalance, immune checkpoint activation, and ultimately the collapse of antitumor immunity, shaping an immune-privileged tumor microenvironment.
PDGFRA-related vaccine development: foundations and prospects
Challenges in glioma immunotherapy
The cardinal challenges in glioma immunotherapy comprise tumor heterogeneity-induced antigen escape and the profoundly immunosuppressive tumor microenvironment (71–73). Despite breakthrough advancements with immune checkpoint inhibitors (ICIs) and chimeric antigen receptor T cell (CAR-T) therapies across diverse solid malignancies, their efficacy remains substantially limited in glioma treatment (74). This therapeutic limitation is primarily attributable to several factors: pronounced molecular and cellular heterogeneity in gliomas, resulting in differential antigen expression profiles that complicate specific tumor antigen identification (75). Elevated expression of immunosuppressive molecules and enrichment of suppressive cellular populations within the glioma microenvironment, impeding effector T cell functionality (76, 77). And tumor cell immune evasion strategies, including downregulation of major histocompatibility complex (MHC) molecules and inhibition of antigen presentation mechanisms (78, 79).
The blood-brain barrier (BBB) further exacerbates the challenges in glioma immunotherapy. The BBB functions as a natural barrier for the central nervous system, stringently restricting the penetration of exogenous substances and immune cells into the brain parenchyma (80). Conventional immunotherapeutic agents encounter significant difficulty effectively traversing the BBB to reach tumor sites and exert their therapeutic effects (81). While disruption of BBB integrity may partially enhance drug penetration, intratumoral hypertension and cerebral oedema further constrain drug permeation into the tumor (82).
The high diversity and redundancy of tumor driver genes and signaling pathways constitute additional obstacles to glioma immunotherapy. Beyond PDGFRA, multiple critical genes—including EGFR, IDH1/2, TERT, PTEN, and TP53—play significant roles in glioma development and progression (44). These genetic mutations or copy number variations frequently co-exist and demonstrate complex regulatory interrelationships (75). PDGFRA functions not merely as an independent oncogenic driver but occupies a pivotal position within the regulatory network of multiple genetic abnormalities. Targeted intervention against PDGFRA may potentially disrupt tumor progression mechanisms driven by cooperative multi-gene mutations, representing a significant breakthrough opportunity for precision therapy in glioma.
Glioma stem cells (GSCs) present another significant challenge. GSCs play crucial roles in glioma initiation, progression, recurrence, and therapeutic resistance (83). These cells maintain their stemness through diverse mechanisms, including activation of signaling pathways such as PDGFRA, Notch, and Wnt, while establishing specialized metabolic and epigenetic regulatory environments (30, 84). Consequently, effective elimination of GSCs or induction of their differentiation represents a key determinant for enhancing immunotherapeutic efficacy.
Addressing these challenges, PDGFRA signaling pathway-related vaccine development represents a significant research direction for confronting the therapeutic dilemmas in glioma treatment. On one hand, the high-frequency mutations and specific expression of PDGFRA provide ideal targets for vaccine design (17, 33). Vaccines targeting PDGFRA mutation epitopes must effectively induce specific T cell responses while simultaneously activating both CD4^+^ and CD8^+^ T cells, potentially overcoming barriers imposed by tumor heterogeneity (45, 85). On the other hand, the close regulatory relationship between PDGFRA and PD-1/PD-L1 pathways provides a rational theoretical foundation for combination interventions with immune checkpoint inhibitors (60). Furthermore, PDGFRA’s association with tumor metabolism, angiogenesis, and GSC stemness maintenance offers additional possibilities for multi-target combination vaccine designs (86). Consequently, the development of optimal PDGFRA vaccines holds promise for surmounting multiple barriers in glioma immunotherapy.
Basic principles of tumor vaccine design
Vaccine therapy represents a transformative breakthrough in glioma treatment, necessitating consideration of multiple factors including target antigen determination, vaccine platform selection, delivery systems, and adjuvant utilization. Diverse vaccine modalities present distinct advantages and limitations, encompassing peptide vaccines, dendritic cell (DC) vaccines, and DNA/RNA vaccines, among others.
Ideal tumor vaccines require highly immunogenic antigens that are exclusively expressed in tumor cells and essential for their survival (87). PDGFRA vaccine design focuses on recognizing and targeting tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs) in glioma cells to stimulate immune responses directed against neoplastic cells (88–90). This process relies on efficient delivery systems (such as mRNA lipid nanoparticles, viral vectors, or dendritic cell loading techniques) to present antigens to immune cells, augmented by adjuvants (such as TLR agonists) to enhance immune activation, thereby improving immune system recognition and killing capacity against tumor cells. Research indicates that while mRNA vaccines have demonstrated efficacy across various malignancies, progress in glioma applications has been comparatively gradual (91).
Currently, the application of personalized mRNA vaccines in GBM treatment is being actively explored (92), providing novel directions for the integration of PDGFRA-targeted vaccines. While mRNA vaccine applications remain in the experimental phase, their preliminarily confirmed immune-activating potential may provide additional theoretical support and practical guidance for PDGFRA vaccine design through in-depth analysis of glioma-specific immune response mechanisms. Future developments hold promise for more targeted and efficacious therapeutic regimens (93).
PDGFRA vaccine design and optimization
PDGFRA vaccine design necessitates consideration of multiple factors, including personalized antigen selection, multi-antigen targeting, and refinement of vaccine platforms or adjuvants—elements that constitute critical determinants for enhancing therapeutic efficacy in glioma vaccination strategies.
Antigen selection constitutes the core element of vaccine design. PDGFRA mutations or aberrant expression in gliomas can generate immunogenic neoantigens—tumor-specific antigens capable of immune system recognition and induction of specific T cell responses (94–97). Ideal PDGFRA vaccine neoantigens should demonstrate high expression in tumor cells while maintaining specificity for individual subjects or subject subgroups receiving the antigen target, thereby reducing off-target toxicity risks and enhancing immunotherapeutic specificity (98, 99). Implementation of this strategy depends on the integrated application of multi-omics technologies (genomics, transcriptomics, and immunopeptidomics).
The development of high-throughput sequencing technologies has provided powerful tools for neoantigen identification. Comprehensive identification of tumor-specific mutations can be achieved through genomic and transcriptomic sequencing of neoplastic cells (100). Furthermore, bioinformatic analyses can predict the binding capacity of mutation-derived peptides to patients’ major histocompatibility complex (MHC) molecules, evaluating their potential for T cell recognition (101, 102).
Delivery systems play a crucial role in vaccine efficacy, not only protecting antigens from degradation but also effectively delivering them to antigen-presenting cells, thereby efficiently activating immune responses. Delivery systems can influence immune reactions through multiple mechanisms, including enhancement of antigen uptake and cross-presentation, as well as activation of inflammatory signaling pathways. Currently, various delivery systems are under investigation, including viral vectors, liposomes, and polymeric nanoparticles, each possessing unique advantages and limitations.
Among these, nanoparticles have emerged as promising tools in cancer therapy, demonstrating significant research potential in targeted drug and gene delivery. These tools can influence immune cells through various mechanisms, such as facilitating targeted delivery and promoting endosomal escape, thereby enabling release of drugs, antigens, and adjuvants at intended targets (103–105). Surface modification of nanoparticles with targeting ligands can further optimize delivery to lymphoid organs or antigen-presenting cells, while their unique physicochemical properties can enhance dendritic cell recognition and uptake (106). The utilization of nanoparticle systems for delivering immunotherapeutic agents is being actively explored, potentially establishing them as effective options for future PDGFRA tumor vaccine therapies. To further enhance vaccine immunogenicity, the application of adjuvants is essential.
Furthermore, adjuvants—key substances that enhance immune responses—are typically used in conjunction with antigens to augment the durability and intensity of immune reactions (107, 108). Adjuvants function through multiple mechanisms, including prolonging antigen exposure, promoting activation of antigen-presenting cells, and inducing cytokine release, ultimately enhancing adaptive immune responses (109). Concurrently, appropriate adjuvant selection warrants consideration to avoid excessive immune system activation, which may precipitate inflammation or other adverse reactions (110). Commonly employed adjuvants encompass aluminum salts, incomplete Freund’s adjuvant (IFA), Toll-like receptor (TLR) agonists, and cytokines (111, 112). Richard G. Everson et al. investigated the efficacy of TLR agonists combined with autologous tumor lysate-pulsed dendritic cell (ATL-DC) vaccines in malignant glioma patients (NCT01204684), demonstrating safety and enhanced systemic immune responses, evidenced by increased interferon gene expression and alterations in immune cell activation (113).
Preclinical research, clinical status, and translational challenges
Preclinical research
PDGFRA plays a pivotal role in glioma pathobiology, with its overexpression and activation representing critical drivers of glioma cell proliferation, angiogenesis, and invasion (114–116). Preclinical investigations have established a robust foundation for elucidating the complex mechanisms through which PDGFRA functions within these neoplasms (Table 1).
Hongyan Zou et al. generated malignant glioma murine models via knockin of mutant PDGFRA, based on cell-autonomous activation of PDGFRA in oligodendrocyte precursor cells (OPCs) (117). Another research team constructed high-grade glioma (HGG) murine models through intracranial implantation of p53-deficient primary astrocytes transduced with PDGFRA D842V mutation (118). Investigations have demonstrated that PDGFRA can induce activation of downstream signaling pathways, including PI3K/AKT and RAS/RAF/MAPK cascades, which play crucial roles in cellular growth, survival, and migration (116, 119–121). Recent studies by Soniya Bastola et al. revealed that endothelial-secreted Endocan (ESM1) enhances glioma proliferation, migration, and angiogenic capacity through direct binding and activation of PDGFRA, subsequently activating PI3K and ERK1/2 pathways (42).
Systematic assessment of current clinical status
Research progress of current glioma vaccines
Current vaccine research conducted across varying pathological subtypes of gliomas, including glioblastoma, astrocytoma, and diffuse glioma, has yielded a series of breakthrough achievements (Tables 2, 3).
Among these, peptide vaccines represent a classical type in glioma vaccination. In a phase II clinical trial of the Survivin-targeted vaccine SurVaxM (NCT02455557), GBM patients who failed the Stupp protocol achieved a median overall survival (mOS) of 25.9 months when combined with temozolomide adjuvant therapy, with both methylated and unmethylated GBM patients demonstrating significant clinical benefit (122). Michael Platten’s team conducted a multicenter phase I clinical study (NCT02454634) of an IDH1(R132H)-targeted peptide vaccine, demonstrating that specific peptide vaccination (IDH1-vac) for grade III-IV (WHO classification) gliomas yielded 3-year progression-free and mortality-free rates of 63% and 84%, respectively (123). Rindopepimut (CDX-110), a peptide vaccine targeting the EGFRvIII mutation, showed survival benefits in several phase II clinical trials for GBM patients (124–126), yet a subsequent phase III trial (NCT01480479) combining CDX-110 with chemotherapy failed to demonstrate significant clinical benefit (mOS 20.1 months vs. 20.0 months) (127).
Given GBM’s pronounced heterogeneity, vaccines targeting standardized single antigens may fail to produce durable antitumor effects. Consequently, multi-antigen vaccines targeting multiple tumor antigens and personalized vaccines theoretically achieve more effective tumor cell elimination. A phase II single-arm trial by Andrew T. Parsa’s research team evaluated the HSPPC-96 vaccine’s safety and efficacy in recurrent GBM (rGBM) patients, demonstrating an mOS of 42.6 weeks (128); their further research indicated this vaccine improved survival rates in newly diagnosed GBM (nGBM) patients receiving standard of care (SOC), with an mOS of 23.8 months (129). The dendritic cell vaccine DCVax-L has shown significant clinical benefit in glioblastoma treatment, with a phase III clinical trial (NCT00045968) demonstrating that, compared to external controls, DCVax-L treatment extended mOS in both nGBM patients (19.3 vs. 16.5 months) and rGBM patients (13.2 vs. 7.8 months) (130).
The research landscape concerning PDGFRA-driven immune evasion in gliomas has yielded significant successes, primarily rooted in the comprehensive characterization of PDGFRA’s oncogenic role. Preclinical models have unequivocally demonstrated PDGFRA’s pivotal involvement in fueling glioma proliferation, angiogenesis, and invasion, further elucidated through the identification of key downstream signaling pathways, including PI3K/AKT and RAS/RAF/MAPK, which are critical for tumor survival and migration. The discovery of Endocan’s direct interaction and activation of PDGFRA offers a novel target for enhancing glioma progression and angiogenic capacity. These foundational discoveries have spurred the development of diverse glioma vaccine platforms, ranging from peptide and tumor-associated antigen vaccines to DNA/RNA and dendritic cell vaccines, many of which have progressed into clinical trials across various stages of glioma. Encouragingly, some of these vaccine strategies have shown preliminary clinical efficacy, with certain cohorts demonstrating potential for extended overall survival when used in combination therapies or in specific patient populations. However, the field is also marked by considerable failures and limitations. The pivotal phase III trial failure of the EGFRvIII-targeted peptide vaccine Rindopepimut (CDX-110) serves as a stark reminder of the translational chasm. Furthermore, the profound heterogeneity inherent in glioblastoma necessitates multifaceted approaches, as single-antigen targeting has proven insufficient for eliciting durable anti-tumor immune responses, and issues of inadequate immunogenicity remain a concern. The ultimate clinical validation of other promising vaccine candidates, such as DCVax-L and HSPPC-96, is still pending, underscoring the challenges in achieving consistent and robust clinical benefits.
Clinical application of existing PDGFRA-targeted drugs
PDGFRA has garnered substantial attention from researchers as a potential therapeutic target in oncology. Various PDGFRA-targeted therapeutic strategies have been explored, including tyrosine kinase inhibitors and monoclonal antibodies (45, 131, 132). The complexity of PDGFRA signaling and its crosstalk with other receptor tyrosine kinases may contribute to resistance against PDGFRA inhibitors, reflecting limited efficacy across multiple clinical trials. Current research indicates that previous tyrosine kinase inhibitors targeting PDGFRA (such as dasatinib and imatinib) demonstrated limited efficacy in clinical trials, potentially due to poor drug tolerance and ineffective blood-brain barrier penetration (133, 134). In contrast, avapritinib has shown preliminary efficacy in HGG patients. Recent studies have preliminarily confirmed avapritinib’s significant potential clinical value in treating pediatric HGG patients carrying specific PDGFRA gene variants (particularly amplification or exon 18 mutations), though validation with additional clinical data is necessary due to small sample sizes. A phase I/II clinical trial (NCT04773782) is currently underway to further evaluate its safety and efficacy (45).
Furthermore, preclinical studies have investigated strategies combining PDGFRA inhibitors with other therapies such as radiotherapy, demonstrating synergistic antitumor effects in certain glioma subtypes (58). Consequently, enhanced understanding of PDGFRA’s role and mechanisms in gliomas could facilitate the development of PDGFRA-related vaccine therapeutic strategies, thereby improving treatment outcomes for patients with this devastating disease.
Bottlenecks in clinical translation
Although avapritinib and other novel tyrosine kinase inhibitors (TKIs) have shown potential in patients with specific PDGFRA mutations, TKI drugs generally face the problems of drug resistance and limited blood-brain barrier penetration efficiency (135). This highlights the necessity of developing therapies with different mechanisms of action, such as therapeutic vaccines. By mobilizing the systemic immune system, vaccines have the potential to generate more durable memory responses and may form synergistic effects with targeted drugs like TKIs.
Translational barriers impeding PDGFRA-targeted vaccines’ progression from research prospects to clinical success encompass multiple practical and technical challenges, including not only the immunosuppressive tumor microenvironment and blood-brain barrier impediments discussed in previous sections, but also complex resistance mechanisms and challenges in real-world technology standardization (136–140).
Glioma cells can develop resistance through multiple pathways, including promotion of epigenetic regulation, activation of bypass signaling pathways, and enhancement of DNA repair capabilities. GBM cells typically exhibit resistance to temozolomide (TMZ), with mechanisms involving O6-methylguanine-DNA methyltransferase (MGMT) expression, mismatch repair gene mutations, and interactions between m6A and histone modifications (141). Additionally, glioma cells can induce resistance through epigenetic modifications, remodeling of the tumor microenvironment, and acquisition of stem cell-like characteristics (137). Activation of multiple signaling pathways also provides opportunities for resistance development. PI3K-AKT, NOTCH, WNT/β-catenin, and MAPK signaling pathways commonly exhibit abnormal activation in gliomas, which not only promotes tumor growth and invasion but also leads to resistance against existing therapeutic approaches (142–145). To overcome this challenge, researchers have explored various synergistic treatment modalities, with combined targeting of multiple signaling pathways considered an effective strategy. Studies have found that EGFR and PDGFRA are frequently co-expressed in gliomas, with EGFR inhibitors affecting PDGFRA signaling through functional transactivation (19). Combined inhibition of EGFR and PI3K/AKT/mTOR pathways can enhance antitumor activity and effectively delay resistance development (146).
Upregulation of immune checkpoint molecules in glioma cells following vaccine therapy may constitute one reason for vaccine failure (147, 148). To counter such immunosuppressive mechanisms, immune checkpoint inhibitors (ICIs) represent ideal adjuncts to vaccine therapy, aiming to overcome potential immunosuppressive or immunologically “cold” tumor microenvironments through inhibition of negative co-stimulatory molecules (such as PD-1/PD-L1) (149, 150). Such combination therapies have demonstrated promising clinical outcomes in other malignancies (including non-small cell lung cancer, melanoma, hepatocellular carcinoma, and bladder cancer), with confirmation in gliomas pending continued investigation (151–153). Preclinical studies have confirmed that PD-1 blockade enhances vaccine-induced immune responses in orthotopic mouse GBM models (149). Additional clinical trials investigating ICI and vaccine combinations for glioma treatment are currently underway (NCT02287428, NCT02960230, NCT04201873).
Glioma heterogeneity imposes stringent requirements for precise antigen presentation and efficient drug delivery, constituting a key technical challenge in tumor vaccine development. Currently, neoantigen vaccines tailored to patient-specific mutations have demonstrated potential to induce effective antitumor immune responses across multiple cancer types (152, 154–157). Concurrently, researchers have developed immune infiltration-related risk models that may facilitate selection of patients suitable for immunotherapy through gene expression analysis, promoting individualized immunotherapeutic approaches for improved clinical outcomes (158). Considering each patient’s tumor’s unique genetic and immunological characteristics, such personalized medical approaches may be crucial for realizing PDGFRA vaccine potential, maximizing therapeutic efficacy while minimizing vaccine toxicity.
Various modern emerging technologies offer new tools for vaccine development and combination strategies. Researchers have developed nanomedicines capable of traversing the BBB, reactivating immunosuppressive TME and triggering anti-tumor immune cascades by activating interferon gene pathways and inhibiting the PD-1/PD-L1 signaling axis (59). Oncolytic viruses, as a novel immunotherapy, exert direct oncolytic effects and enhance antigen exposure, converting “cold” TME to “hot” TME in gliomas, ultimately activating anti-tumor immunity. A phase II clinical trial demonstrated G47Δ (oncolytic herpes virus) efficacy and safety for residual or recurrent glioblastoma, with an 84.2% one-year survival rate and median OS and PFS of 20.2 and 4.7 months, respectively. Additionally, PVSRIPO (recombinant non-pathogenic poliovirus-rhinovirus chimera) demonstrated efficacy in a phase I clinical trial enrolling 61 glioma patients (NCT01491893) (159).
From a clinical translation perspective, PDGFRA-targeted vaccine strategies for glioma offer a compelling duality of significant promise and formidable challenges. The inherent capacity of immunotherapies, particularly vaccines, to overcome glioma’s pronounced cellular and molecular heterogeneity through multi-antigenic or personalized approaches presents a distinct advantage, potentially eliciting more sustained and robust anti-tumor immune responses than conventional monotherapies. This approach also holds the promise of establishing durable immunological memory, crucial for long-term disease control. Furthermore, the synergistic potential of integrating vaccine therapies with existing treatments like chemotherapy and emerging immune checkpoint inhibitors offers a powerful avenue to augment overall therapeutic efficacy. Notably, many vaccine candidates have demonstrated favorable safety profiles in early-stage investigations, a critical factor for patient acceptance and broader clinical adoption.
However, the path to widespread clinical translation is impeded by substantial obstacles. The blood-brain barrier (BBB) remains a significant physical impediment, limiting the efficient delivery and in situ activation of vaccine components within the tumor microenvironment. This challenge is exacerbated by the profoundly immunosuppressive nature of the glioma milieu, which actively impairs anti-tumor immune cell function and persistence, presenting a critical hurdle for ensuring effective antigen presentation and robust cytotoxic T lymphocyte activation. The development and standardization of clinical-grade manufacturing processes, particularly for personalized vaccines, introduce substantial logistical and cost barriers, demanding scalable and reproducible technologies. Moreover, the observed discrepancies between promising preclinical outcomes and clinical trial results underscore the limitations of current animal models, which often fail to fully recapitulate the “cold tumor” characteristics of human gliomas, hindering accurate prediction of in vivo efficacy (160). Consequently, advancing PDGFRA vaccines from bench to bedside necessitates rigorous preclinical evaluation of safety and immunogenicity, alongside optimization of administration methods, dosages, and scheduling, all while navigating complex regulatory landscapes and refining patient selection biomarkers to identify individuals most likely to benefit from these innovative immunotherapeutic interventions.
Future development directions and research priorities
The treatment paradigm for gliomas presents a formidable clinical challenge. Tumor vaccines constitute an effective approach for enhancing therapeutic outcomes in glioma patients; however, compared with other solid tumors, barriers such as cranial biological barriers and the unique tumor and immune microenvironment impact vaccine efficacy. Consequently, researchers have conducted extensive investigations in immunotherapy and precision oncology domains to explore novel methodologies. Currently, select glioma vaccines have demonstrated positive interim clinical research outcomes, though developmental potential remains substantial.
Personalized vaccine strategies based on neoantigens
Glioma PDGFRA vaccines necessitate thorough consideration of tumor immune complexity and inter-patient heterogeneity. Tumor heterogeneity, immune status, and genetic background may influence vaccine efficacy and constrain immune cell infiltration and function. Consequently, personalized vaccine design is progressively emerging as a future developmental trend. Through genomic and immunomic analyses of patient tumors, customized therapeutic strategies adapted to individual tumor characteristics can be provided, acknowledging each patient’s unique tumor biology, thereby substantially enhancing therapeutic outcomes (161, 162). Future development of personalized vaccines targeting patient-specific neoantigens, through genomic identification of mutations and peptide vaccine design, aims to achieve superior tumor regression and anti-tumor responses (163). Additionally, optimization of vaccine administration protocols, including dose selection, delivery route refinement, and rational timing arrangements, constitutes an important approach for improving vaccine efficacy.
Utilizing advanced technologies to optimize vaccine design and delivery
Novel technological developments provide powerful tools for in-depth investigation of glioma vaccines. With advancements in bioinformatics and artificial intelligence, challenges in identifying and targeting novel antigens, including PDGFRA-related antigens, may potentially be resolved (164). Multi-omics technologies comprehensively elucidate molecular characteristics of tumors and immune systems. Through utilization of genomics, transcriptomics, proteomics, and metabolomics, potential vaccine targets can be identified, immune profiles of tumor microenvironments characterized, and vaccine influences on immune cell activity elucidated (165). Furthermore, identification of biomarkers predicting vaccine response, such as PDGFRA status or immune infiltration characteristics, facilitates selection of patients most likely to benefit from vaccine therapy. Development of novel delivery systems constitutes another priority direction. Systems based on nanoparticles, viral vectors, or liposomes capable of specifically targeting glioma cells or tumor microenvironments enhance vaccine capacity to traverse the BBB while reducing off-target effects. Additionally, exploration of ultrasound or other physical techniques to temporarily disrupt the BBB represents a potentially promising strategy for promoting vaccine delivery.
Combined treatment strategies to enhance tumor vaccine efficacy
Immunotherapy resistance constitutes a critical bottleneck constraining glioma vaccine efficacy enhancement, with in-depth analysis of underlying mechanisms and exploration of effective countermeasures representing an important future research direction. Tumor microenvironment complexity significantly influences vaccine efficacy, with abundant immunosuppressive cells and aberrant immune checkpoint activation substantially limiting immune cell infiltration and cytotoxic function (166, 167). Addressing this challenge, combined treatment strategies emerge as crucial breakthrough approaches. Concurrent administration of glioma tumor vaccines with immune checkpoint inhibitors (such as PD-1/PD-L1 inhibitors) effectively blocks immunosuppressive signaling pathways, reactivating T-cell anti-tumor activity and enhancing vaccine-induced immune responses. Simultaneously, PDGFRA-targeted therapies combined with vaccines precisely modulate critical tumor cell signaling pathways, overcoming resistance arising from signaling abnormalities. Designing vaccines targeting multiple antigens constitutes another important strategy preventing immune evasion due to antigen loss. Multi-stage combined therapeutic approaches may provide promising supplementary regimens, with PDGFRA tumor vaccines administered concurrently with chemotherapy, radiotherapy, or immune checkpoint inhibitors potentially further enhancing therapeutic outcomes, overcoming tumor resistance, and achieving more sustained tumor control (168–171) (Figure 4).
PDGFRA-targeted vaccine strategy in glioma immunotherapy: Mechanism, challenges, and future directions. The illustration depicts the design and mechanism of PDGFRA-targeting vaccines (e.g., peptide, mRNA, DC, or viral vector platforms), which utilize antigens such as full-length PDGFRA, extracellular domains, or mutation-specific epitopes to activate APCs and induce cytotoxic (CD8+) and helper (CD4+) T-cell responses, ultimately leading to tumor cell killing and potential remodeling of the immunosuppressive TME. Key clinical challenges include the blood-brain barrier, tumor heterogeneity, immune-suppressive TME, and limited immunogenicity of PDGFRA as a self-antigen. Future optimization may involve combinatorial approaches (e.g., immune checkpoint inhibitors, chemotherapy), nano-delivery systems to bypass the BBB, personalized neoantigen strategies, and TME-modulating therapies to enhance efficacy.
Conclusion
PDGFRA-mediated signaling networks establish complex interactions between glioma cells and the immunosuppressive microenvironment, constituting a core barrier to immunotherapeutic efficacy. Advances in multi-omics technologies and neoantigen identification are facilitating development of highly specific, individualized PDGFRA-targeted vaccine designs. Combined with next-generation delivery systems and effective adjuvants, such vaccines may induce robust and durable anti-tumor immune responses, potentially facilitating transformation of gliomas from “immunologically cold” to “hot” tumors. Prospectively, deep integration of artificial intelligence, molecular typing, nanotechnology, and immune engineering will play critical roles in overcoming glioma immune evasion and achieving precise immunotherapy, offering new therapeutic prospects for patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Weller M Wen PY Chang SM Dirven L Lim M Monje M . Glioma. Nat Rev Dis Primers. (2024) 10:33. doi: 10.1038/s 41572-024-00516-y, PMID: 38724526 · doi ↗ · pubmed ↗
- 2Sharma P Aaroe A Liang J Puduvalli VK . Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol Adv. (2023) 5:vdad 009. doi: 10.1093/noajnl/vdad 009, PMID: 36968288 PMC 10034917 · doi ↗ · pubmed ↗
- 3Yabo YA Niclou SP Golebiewska A . Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro Oncol. (2022) 24:669–82. doi: 10.1093/neuonc/noab 269, PMID: 34932099 PMC 9071273 · doi ↗ · pubmed ↗
- 4Paugh BS Zhu X Qu C Endersby R Diaz AK Zhang J . Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. (2013) 73:6219–29. doi: 10.1158/0008-5472.Can-13-1491, PMID: 23970477 PMC 3800209 · doi ↗ · pubmed ↗
- 5Rogers MA Fantauzzo KA . The emerging complexity of PDGF Rs: activation, internalization and signal attenuation. Biochem Soc Trans. (2020) 48:1167–76. doi: 10.1042/bst 20200004, PMID: 32369556 PMC 7722362 · doi ↗ · pubmed ↗
- 6Jackson CM Choi J Lim M . Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. (2019) 20:1100–9. doi: 10.1038/s 41590-019-0433-y, PMID: 31358997 · doi ↗ · pubmed ↗
- 7Adhikaree J Moreno-Vicente J Kaur AP Jackson AM Patel PM . Resistance mechanisms and barriers to successful immunotherapy for treating glioblastoma. Cells. (2020) 9:263. doi: 10.3390/cells 9020263, PMID: 31973059 PMC 7072315 · doi ↗ · pubmed ↗
- 8Akiyama T Yasuda T Uchihara T Yasuda-Yoshihara N Tan BJY Yonemura A . Stromal reprogramming through dual PDGFRα/β Blockade boosts the efficacy of anti-PD-1 immunotherapy in fibrotic tumors. Cancer Res. (2023) 83:753–70. doi: 10.1158/0008-5472.Can-22-1890, PMID: 36543251 · doi ↗ · pubmed ↗