Coexisting genetic kidney disease explains many cases of ‘familial’ IgA nephropathy where the proband has biopsy-confirmed mesangial IgA deposits
YuXin Li, Jing Zhang, Zhuo Jun Yin, Ziming Zhou, Bocheng Huang, Khalid Mahmood, Deb Colville, David Barit, Russell Auwardt, Robert Fassett, Kathy Paizis, Francesco Ierino, Timothy Pianta, David Langsford, Mary Huang, Judy Savige

TL;DR
This study finds that many cases of familial IgA nephropathy are actually due to a coexisting genetic kidney disease, not just IgA nephropathy alone.
Contribution
The study shows that genetic kidney disease often coexists with IgA nephropathy in families, explaining some cases previously labeled as purely familial.
Findings
45% to 82% of families with familial IgA nephropathy had genetic kidney disease variants.
Some probands lacked the genetic variant found in affected family members.
Two sporadic IgA nephropathy cases also had genetic kidney disease variants.
Abstract
One in seven people with IgA nephropathy has another apparently-affected family member. This study examined how often biopsy-proven familial and sporadic IgA nephropathy were associated with genetic kidney disease. Eleven unrelated people with biopsy-proven IgA nephropathy and another family member with kidney tests compatible with IgA nephropathy were recruited. All available family members were assessed for genetic kidney disease, using Whole Exome Sequencing (WES). Their results were compared with those of 39 people with sporadic IgA nephropathy. All sequencing results were filtered for pathogenic variants in genes associated with genetic kidney disease (Genomics England panels, n = 384). Variants were assessed for pathogenicity using ClinVar and the ACMG/AMP criteria in Alamut. Nine of the 11 probands (82%) with familial nephropathy and 30 of those with sporadic disease (77%) had…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
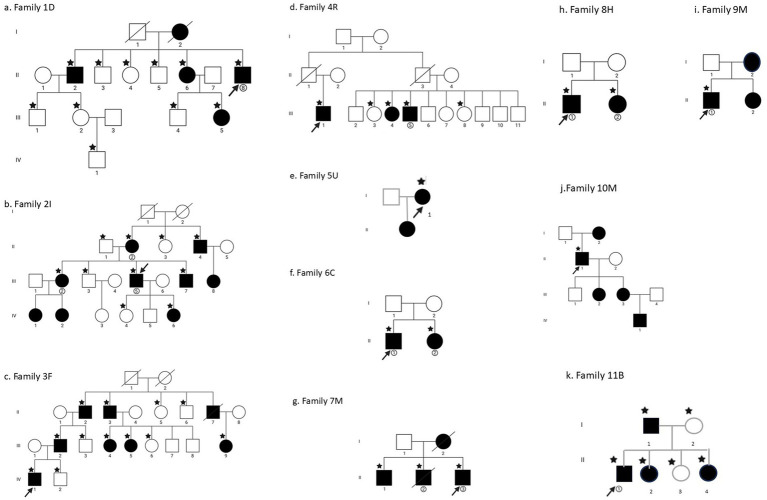 Figure 1
Figure 1| Family | Gene | Disease and mode of inheritance | Sequence change | Alleles in gnomAD (v.2.1) | Alamut | ClinVar |
|---|---|---|---|---|---|---|
| 1D proband |
| AR predisposes to bacterial infections and autoimmune disease | 247 | Pathogenic (PVS1, PM2, PP5) score = 11 | Pathogenic/ | |
| 2I proband |
| AD, Alport syndrome | None | Likely pathogenic (PM1, PM2, PP3, | Pathogenic/ | |
| 3F proband |
| XL Alport syndrome | None | Pathogenic (PVS1, PM2) | Not found | |
| Other members of 3F |
| AD Alport syndrome | 17 | Pathogenic (PVS1, PM2, PP5), | Pathogenic **; rs35138315 | |
| 7M proband |
| AD, HNF1B-nephropathy | None | VUS (PM2, PP5) | Likely pathogenic* | |
| 8H proband |
| AD, Charge syndrome | 16 | VUS (PM1, PM2, PP3) score = 5 | Conflicting | |
| Other members of 11B |
| FSGS | 23 | Pathogenic (PVS1, PM2) | Conflicting* (VUS/Benign); Likely pathogenic46; rs758452999 | |
| Families where only the proband was tested | ||||||
| 5U proband |
| AD Alport syndrome | 5 | VUS (PM2, PP3, PP5) score = 4 | Pathogenic/ | |
| 6C proband |
| XL, Dent disease | None | VUS (PM1, PM2, PP3) | Not found; not in HGMD | |
| 9M proband |
| AD Alport syndrome | 7,694 | VUS (PM1, PM2, | VUS*; rs1800516 | |
|
| FSGS (risk factor) | 4,448 | VUS (PM1, BP4) | Conflicting (P, LP, VUS, B); | ||
| 10M proband |
| XL Alport syndrome | 16 | Likely pathogenic (PM1, PM2, PP3, | Pathogenic/ | |
| Individuals with sporadic IgA nephropathy ( | ||||||
| S1 |
| AD Alport syndrome | 2 | Pathogenic (PM1, PM2, PP3, PP5), score = 6 | P/LP **; | |
| S47 |
| Cystinuria (AR and AD) | 24 | Likely pathogenic (PM1, PM2, PP3, PP5) | P/LP**; | |
| Individuals with IgA vasculitis ( | ||||||
| No pathogenic variants detected in genes studied | ||||||
| Individuals with other forms of glomerulonephritis ( | ||||||
| G19 |
| Distal renal tubular acidosis type 1, AD | 10 | Likely pathogenic (PM1, PM2, PP3, PP5) score = 6 | Pathogenic**’ rs121912748 | |
| Family | Proband (sex, age, ancestry) | Clinical features in the proband with biopsy-proven IgA glomerulonephritis | Total number of affected family members who were genetically tested | Number of first-degree family members sequenced | Pathogenic variant in the proband in the genetic kidney disease gene | Pathogenic variant in affected family members |
|---|---|---|---|---|---|---|
| 1D | M, 46, N European | Hematuria, proteinuria, kidney failure, transplant | Four (brother also with biopsy-proven IgA disease, sister, niece) | Eleven | C9:p.Cys 24Ter | C9 variant in proband, three affected, and one unaffected family member |
| 2I | M, 60, N European | Hematuria, proteinuria | Six (mother, uncle, cousin, sister, brother, daughter). Sister has a kidney transplant | Ten | p.Gly395Glu in | |
| 3F | M, 31, N European | Hematuria, impaired kidney function | Seven (father, grandfather, great uncle, three second cousins). Great uncle on dialysis | Twelve | p.Pro1384ArgfsTer39 in | |
| 4R | M, 60, N European | Hematuria, proteinuria, impaired kidney function, and IgA vasculitis as a child | Three (proband and two cousins) | Five | None | None |
| 5U | F, 64, S European | Hematuria, nearly normal kidney function | One (daughter also affected) | One | p.Gly466Arg in | Family members were not examined |
| 6C | M, 51, N European | Kidney failure, transplant | One (sister affected mildly) | One | p.Arg537Gln in | Family members were not examined |
| 7M | M, 38, S European | Hematuria, impaired kidney function | Proband and two brothers (both on dialysis) | Three | p.Pro437Leu in | |
| 8H | M, 56, N European | Hematuria, kidney failure, and a kidney transplant | Two (a sister also had biopsy-proven IgA glomerulonephritis) | Two | p.Arg1100His in | |
| 9M | M, 55, N European | Hematuria, impaired kidney function | One (sister and mother also had hematuria) | One | p.Gly545Ala in | Family members not examined |
| 10M | M, 68, S European | Hematuria, kidney failure at 60, kidney transplant | One (mother, two daughters, and a grandson) was also affected | One | p.Gly624Asp in | Family members were not examined |
| 11B | M, 35, N European | Hematuria, proteinuria, kidney failure, and kidney transplant | Four (father, two sisters) | Six | None |
| Family and family member | Hematuria | Proteinuria | Impaired kidney function | Renal biopsy | Pathogenic variant |
|---|---|---|---|---|---|
| Family 1D | |||||
| II-2 | YES | IgA gn | p.Cys54Ter in | ||
| II-6 | YES | YES | YES | p.Cys54Ter in | |
| II-8 (proband) | YES | YES | YES (eGFR<20 mL/min/1.73 m2), Transplant | IgA gn | p.Cys54Ter in |
| III-5 | YES | NO | NO | p.Cys54Ter in | |
| Family 2I | |||||
| II-2 | YES | YES | p.Gly395Glu in | ||
| II-4 | YES | YES | p.Gly395Glu in | ||
| III-2 | YES | YES | YES, Transplant | IgA gn | p.Gly395Glu in |
| III-5 (proband) | YES | NO | IgA gn | p.Gly395Glu in | |
| III-7 | YES | p.Gly395Glu in | |||
| IV-6 | YES | YES | NO | p.Gly395Glu in | |
| Family 3F | |||||
| II-2 | YES | YES | None detected | ||
| II-3 | YES | YES | YES, Dialysis | p.Ser969Ter in | |
| III-2 | YES | FSGS | None detected | ||
| III-4 | YES | YES | p.Ser969Ter in | ||
| III-5 | YES | p.Ser969Ter in | |||
| III-9 | YES | None detected | |||
| IV-1 (proband) | YES | YES | IgA gn | p.Pro138ArgfsTer39 in | |
| Family 4R | |||||
| III-1 (proband) | YES | YES | YES | IgA vasculitis, Transplant | None detected |
| III-4 | YES | None detected | |||
| III-5 | YES | None detected | |||
| Family 5U | |||||
| I-1 (proband) | YES | NO | NO | IgA gn | p.Gly466Arg in |
| Family 6C | |||||
| II-1 (proband) | YES | YES | YES, Transplant | IgA gn | p.Arg537Gln in |
| II-2 | YES | YES | NO | p.Arg537Gln in | |
| Family 7M | |||||
| II-1 | YES | YES | YES, Dialysis | Diabetes, kidney cysts | p.Pro437Leu in |
| II-2 | YES | YES | YES, Dialysis | FSGS, diabetes, and kidney cysts | p.Pro437Leu in |
| II-3 (proband) | YES | YES | YES | IgA gn, diabetes, kidney cysts | p.Pro437Leu in |
| Family 8H | |||||
| II-1 (proband) | YES | YES | YES, Transplant | IgA gn | p.Arg1100His in |
| II-2 | YES | NO | NO | IgA gn | None detected |
| Family 9M | |||||
| II-1 (proband) | YES | NO | NO | IgA gn | p.Gly545Ala in |
| II-2 | YES | Not tested | |||
| Family 10M | |||||
| II-1 (proband) | YES | YES | YES, Transplant | IgA gn | p.Gly624Asp in |
| III-2 | YES | Not tested | |||
| III-3 | YES | Not tested | |||
| IV-1 | YES | Not tested | |||
| Family 11B | |||||
| I-1 | YES | YES | YES | None detected | |
| II-1 (proband) | YES | YES | YES, Transplant | IgA gn | None detected |
| II-2 | YES | YES | YES | FSGS | c.*1 + 1G > C in |
| II-4 | YES | None detected | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal Diseases and Glomerulopathies · Genetic and Kidney Cyst Diseases · Vasculitis and related conditions
Introduction
IgA nephropathy is possibly the commonest form of glomerulonephritis worldwide (1, 2). It is characterized by macroscopic hematuria associated with mucosal infections, and diffuse mesangial deposits of IgA, IgG, and C3. One in four of those affected develops kidney failure after 20 years (3), and IgA nephropathy accounts for 6% of patients requiring renal replacement therapy (4).
Our current understanding of the pathogenesis of IgA nephropathy includes ‘multiple hits.’ (5) Mucosal infections, including EBV (6) result in galactose-deficient IgA1 (7), which is targeted by anti-glycan IgG antibodies (8), forming immune complexes in the glomerular mesangium and activating complement by the alternative and lectin pathways (9). The diverse properties of these immune complexes (composition, molecular mass, etc.) may explain the presence or absence of symptomatology.
The evidence for genetic involvement in the pathogenesis of IgA nephropathy is complicated. Fifteen percent of people with biopsy-proven IgA nephropathy have another apparently-affected family member (10), often with disease in each generation. These familial forms of IgA nephropathy are typically more severe than sporadic disease, with earlier onset of kidney failure (11, 12). However, the familial association and increased severity are unexplained.
In IgA nephropathy, the abnormal mesangial IgA1 deposits are determined genetically by variants in the C1GALT1 gene (13). These variants likely contribute to the increased disease susceptibility in people of East Asian and Southern European ancestries (14). However, the effect of the C1GALT1 variants is unclear since only some family members with this change develop IgA nephropathy. Furthermore, while galactose-deficient mesangial IgA1 deposits are noted post-mortem in up to 20% of the population, many fewer people have an active glomerulonephritis (15–17).
Nevertheless, genetic variants that predispose to IgA nephropathy have been identified in potentially relevant pathways in various Genome Wide Association Studies (GWAS). These pathways affect mucosal innate immunity against pathogens (DEFA cluster, PADI4, IRF4, UBR5, CARD9, PSMB8/PSMB9, FCRL3); IgA1 production (TNFSF13, LIF, OSM) and glycosylation (ST6GAL1); antigen presentation (HLA cluster, PSMB8/PSMB9, TAP1/TAP2); lymphocyte maturation (DUSP22, VAV3); and complement activation (CFH, CFHR3-CFHR1, ITGAM, ITGAX) (18–21). HLA and additional genes have been implicated in IgA nephropathy and related diseases such as IgA vasculitis (formerly Henoch-Schonlein purpura) (22), Inflammatory Bowel Disease (23) and Ankylosing Spondylitis (24). In general, common variants in the GWAS-identified genes each have a small additive effect on disease risk, but rare deleterious variants in the same genes may have more pronounced consequences.
Finally, monogenic causes have been reported in some cases of familial IgA nephropathy. Affected genes include SPRY2, which encodes an inhibitor of B cell proliferation (25), as well as complement pathway genes (26, 27), and occasionally other apparently coincidental genetic kidney diseases (28–31). Up to 20% people with familial IgA nephropathy have been reported to have autosomal dominant (AD) or XL Alport syndrome, two of the most common genetic diseases affecting the kidney (32). Sometimes immunodeficiency genes are also associated with the development of autoimmune glomerulonephritis (33, 34).
This study examined how often familial causes were associated with an underlying genetic kidney disease, and differed from previous studies in that multiple family members were examined. These results were compared with those from people with sporadic IgA nephropathy.
Participants and methods
Participants
All 11 probands referred consecutively with familial IgA nephropathy from teaching hospitals between 2014 and 2022 were studied. Each family included at least one member with biopsy-proven disease and another person considered affected because of hematuria (>trace) or proteinuria (≥1+) on dipstick of freshly collected urine specimens (Ames, Germany), impaired kidney function (eGFR < 90 mL/min/1.73 m^2^) without another obvious cause, or biopsy evidence of IgA nephropathy. End-stage Kidney failure (eGFR<15 mL/min/1.73 m^2^; dialysis-dependence or a current kidney transplant) was noted. The renal biopsies themselves were not available for study.
Furthermore, 39 consecutive unrelated patients with biopsy-proven IgA nephropathy but without another affected family member with IgA nephropathy or kidney disease (‘sporadic’ cases) were also examined. People with a secondary cause of IgA nephropathy (e.g., liver disease, etc.) were excluded.
A further five people with biopsy-proven IgA vasculitis and nine with sporadic glomerulonephritis [lupus nephritis in eight; Focal and Segmental glomerulosclerosis (FSGS) in one] were also studied.
This project was approved by the Austin Health Human Research Ethics Committee (HREC/16/Austin/538) in accordance with the principles of the Declaration of Helsinki, and all participants provided written informed consent for their involvement.
Whole exome sequencing and variant filtering
Participants provided peripheral blood from which DNA was extracted using a QIAamp DNA Blood Mini Kit (Qiagen, Netherlands) and 50 ng of purified DNA underwent Whole Exome Sequencing (WES) using libraries prepared with the SureSelect XT Clinical Research Exome v2 target enrichment kits (Agilent Technologies, CA, USA) on an Illumina NovaSeq platform (California) at the Australian Genome Research Facility, Melbourne. Germline single-nucleotide variants (SNV) and short insertions/deletions (indels) were called using standard workflows based on the Genome Analysis Toolkit (GATK v4.4.0) (35). The variants were annotated using the Ensembl Variant Effect Predictor tool (36).
Variants were filtered for changes in 384 genes from the Genomics England panels for genetic kidney disease (Hematuria, Renal proteinuria, CAKUT Cystic kidney disease, and Renal ciliopathies, Kidney failure with no obvious cause) (Supplementary Table S1).1 HLA variants were not examined.
Genetic variants were filtered to identify rare variants [<30 alleles in gnomAD v2.1.1 (37)] that were pathogenic or likely pathogenic according to ACMG/AMP criteria (38) in Alamut2 based on the in silico prediction tools, Polyphen2, SIFT, and Mutation Taster (39–41), or pathogenic or likely pathogenic in ClinVar.3 The scores provided were calculated automatically by Alamut. Scores of at least 5, or those classified as likely pathogenic or pathogenic in ClinVar, were also noted. Two pathogenic variants in the same person were required for the diagnosis of a biallelic kidney disease.
Results
Familial IgA nephropathy (11 families)
Eleven probands with familial IgA nephropathy and seven members of their families (median 4, range 2–9) were studied clinically and underwent WES (median 3, range 1–12) (Tables 1–3; Figures 1a–k). Nine families (82%) had at least one member with end-stage kidney failure.
Family trees of the patients studied here. (a–c) Families 1–3; (d–g) Families 4–7; (h–k) Families 8–11. The index case is indicated by an arrow, and all family members are indicated by generation and birth order. Those with features of kidney disease (hematuria, proteinuria, and impaired kidney function) are demonstrated by the filled squares or circles. Those with a star underwent whole exome sequencing.
Of the 11 families studied, five (45%) had pathogenic variants previously associated with monogenic kidney diseases (2I, 3F, 5U, 7M, and 10M) (Tables 1, 2). Alport syndrome was found in the probands of four of these families (2I, 3F, 5U, and 10M), and the other had an HNF1B pathogenic variant and HNF1B-nephropathy.
Four further families (6C, 8H, 9M, and 11B) had interesting and possibly pathogenic variants, one additional family had two risk factors for kidney failure (9M), and one (1D) had a heterozygous variant in a gene (C9) previously associated with IgA nephropathy in the biallelic form.
Two of the families (3F and 11B) had members other than the proband who had a pathogenic (3F, p.Ser969Ter in COL4A4) or possible pathogenic variant (11B, c.1 + 1G > C in INF2) in genes affected in kidney disease (AD Alport syndrome, FSGS, respectively). Thus, this second variant in family 3F affected an Alport gene.
All disease-causing variants were heterozygous or hemizygous, rather than biallelic. Only two families (1D and 4R) had no suspected genetic kidney disease at all in the proband or a family member.
Families with genetic kidney disease
Five families had disease-causing variants. Four of the families had disease-causing variants in the Alport genes. Three families (2I, 5U, and 10M) each had one pathogenic variant in an Alport gene, and one (3F) had two pathogenic variants in Alport genes. The affected genes were COL4A5 in two families (X-linked Alport syndrome), and COL4A3 and COL4A4 (AD Alport syndrome) in the others.
Family 3F had p.Pro1384ArgTer39 in COL4A5 in the proband only, and three other 3F family members had a different heterozygous pathogenic variant (p.Ser969Ter in COL4A4) that was not present in the proband but segregated with hematuria. No family member had both variants. Proband 5U had a pathogenic variant in COL4A4 (p.Gly466Arg) an Alamut score of 4, but which was considered Pathogenic in ClinVar. Furthermore, Proband 9M had the p.Gly545Ala variant in COL4A4, which may be associated with hematuria and kidney failure (42), but is generally not considered pathogenic. It was found together with an NPHS2 variant (p.Arg229Gln) that results in FSGS in a biallelic disease and has been reported previously to worsen the effect of a monoallelic COL4A3 or COL4A4 variant (43, 44). Both the p.Gly545Ala in COL4A4 and p.Arg229Gln in NPHS2 are considered ‘risk factors’ for kidney failure but are not on their own associated with hematuria or proteinuria, respectively.
Furthermore, proband 7M had a heterozygous variant p.Pro437Leu in HNF1B, which, despite a pathogenicity score of 3 in Alamut, was considered likely pathogenic in ClinVar and consistent with the clinical phenotype of HNF1B-nephropathy in the patient and his two affected brothers, who all had diabetes, kidney cysts, infertility, and kidney failure.
Families with possibly disease-causing variants
Proband 1D had a heterozygous pathogenic variant in C9, which has been associated previously in the biallelic form with IgA nephropathy (26). This variant was also found in three other affected and one unaffected family member. While there is currently no known role for the monoallelic form of this variant in the pathogenesis of IgA nephropathy, at least one person has been reported with a low but detectable C9 level (45). This suggests a heterozygous variant that was not confirmed genetically.
Proband 6C had a possible variant in CLCN5 (Dent disease), with a pathogenicity score of 5 in Alamut, and which was classified as a VUS. Inheritance for this gene is X-linked. The proband had developed kidney failure in his forties and his sister had hematuria and proteinuria, with mildly impaired kidney function but had not undergone WES. This variant was not present in ClinVar, LOVD, or HGMD. No more clinical details were available.
Proband 9M had variants in COL4A4 and NPHS2 (p.Gly545Ala, p.Arg229Gln, respectively) that are generally considered ‘risk factors’ for kidney failure rather than disease-causing, but the effect of the combination is unknown.
Proband 8H had a possibly pathogenic variant (p.Arg1100His) in the CHD7 gene for Hypogonadotrophic hypogonadism 5 with or without anosmia/ CHARGE syndrome (Coloboma, Heart defects, Choanal atresia, Growth restriction, Genital abnormalities, and Ear abnormalities), which had a pathogenicity score of 5 in Alamut and was classified as a VUS. The patient had reflux nephropathy and low testosterone levels consistent with the variant being pathogenic, but none of the other facial, ocular, or cardiac features of CHARGE syndrome.
The proband in family 11B who had a kidney transplant had no pathogenic variant demonstrated but his sister with proteinuria had the c.*1 + 1G > C variant in INF2 that was assessed as Pathogenic with a score of 10 in Alamut and as Conflicting in ClinVar (VUS and Benign). A review of genetic causes of the corresponding disease, Charcot–Marie–Tooth disease/FSGS, classified this variant as likely pathogenic (46). The sister’s clinical and renal biopsy features were consistent with FSGS.
Sporadic IgA nephropathy (n = 39)
Thirty of the 39 individuals (77%) with sporadic IgA nephropathy had kidney failure, six (15%) had normal kidney function, and three (8%) had unknown kidney function.
Two (5%) had a heterozygous pathogenic variant consistent with genetic kidney disease (Table 3). These diseases were AD Alport syndrome (p.Gly619Arg in COL4A3); and AD Cystinuria (p.Arg333Trp in SLC7A9, Pathogenic/Likely pathogenic in ClinVar).
IgA vasculitis (n = 5)
These included four individuals with childhood—and one with adult-onset disease, two of whom had kidney failure (40%). None had a pathogenic variant in a gene affected in genetic kidney disease (Table 3).
Other autoimmune forms of glomerulonephritis (n = 9)
These included eight individuals with SLE and one with FSGS, where four (44%) had kidney failure, three (33%) had normal kidney function, and kidney function was unknown in two (22%). One person (11%) with normal kidney function had a heterozygous pathogenic SLC4A1 variant consistent with the diagnosis of AD Distal renal tubular acidosis type 1, and there were no other genetic kidney diseases (Table 3).
Discussion
This study demonstrated that families with IgA nephropathy commonly have an underlying genetic kidney disease that explains the familial occurrence. At least five, and probably more, of the 11 families described here had a pathogenic variant in a kidney disease-causing gene. Four families had pathogenic variants in the Alport genes, and five different Alport variants were detected. Genetic kidney disease was also found in 5% of individuals with sporadic IgA nephropathy, which was much less common than in the families.
Genetic kidney disease was found more often in this study than reported previously, in part because family members were tested and sometimes had a pathogenic variant not present in the index case. Our cohort was unbiased since the probands were those referred consecutively with familial IgA nephropathy over the period of review. Underlying genetic kidney disease in familial IgA nephropathy is likely to be even more common than demonstrated here because WES was used for genetic testing but detects only 80% of pathogenic changes (47–49) and sometimes overlooks large deletions, deep splicing changes and variants in alternative disease-causing genes.
These observations suggest that the mesangial IgA deposits, which are normally present in 20% of the population, coexist with genetic kidney disease (15, 16) and contribute to the more severe clinical course seen in familial IgA nephropathy (11, 12).
The genetic kidney diseases identified in familial IgA nephropathy reflect the commonest seen, namely, AD and XL Alport syndrome, and AD Tubulointerstitial kidney disease (ADTKD) (48). Another common disease, AD Polycystic Kidney Disease, was probably excluded based on family history and renal imaging. However, additional pathogenic variants in ADTKD-HNF1B and ADTKD-MUC1 might have been missed because WES often does not recognize the large deletions that occur in HNF1B and the typical MUC1 frameshift mutations that require targeted testing.
None of the genetic diagnoses demonstrated here were suspected before testing. In particular, the cysts in Family 7M with ADTKD-HNF1B nephropathy were only recognized on reverse phenotyping. Our cohort included two males with XL Alport syndrome, but neither had the typical hearing loss nor ocular abnormalities. The COL4A5 p.Gly624Asp variant in Family 10M is associated with mild disease, late-onset kidney failure, and no extrarenal features. AD Alport syndrome was the commonest genetic diagnosis (32) but also was not associated with hearing loss or ocular abnormalities (50). Although the glomerular basement membrane is typically thinned in AD or lamellated in XL Alport syndrome, these features were not helpful diagnostically in this cohort. This was because electron microscopy of the kidney biopsy is not routinely performed in Australia; and in late stage kidney disease, the glomeruli are often too scarred to identify the membrane changes typical of Alport syndrome.
While each proband in the cohort had biopsy-proven IgA nephropathy, the diagnosis was often made clinically in other family members. In retrospect, the finding of hematuria, proteinuria, or impaired kidney function may have indicated a genetic kidney disease rather than IgA nephropathy. Indeed, this study found that the proband may have had IgA nephropathy but not the genetic disease present in other family members.
Familial IgA nephropathy is typically found in multiple members of a generation and in several generations, consistent with AD or XL inheritance. Our findings of coexisting genetic diseases with AD or XL inheritance were consistent with this. Autosomal recessive (AR) kidney diseases, such as FSGS, and the ciliopathies and tubulopathies, which are generally rarer, and more likely to occur sporadically, were not identified in this cohort. Any sex-based difference in severity may not be obvious with later onset disease and in families comprising mainly boys or affected females (51).
Genetic kidney disease occurred in 5% of our series of sporadic cases of IgA nephropathy, which was much less often than in familial disease. However, many sporadic cases also had kidney failure and a consequently increased risk of genetic disease (52). Coexisting genetic kidney disease was also present in a patient with autoimmune glomerulonephritis, but not in IgA vasculitis.
The prevalence of kidney failure in our cohorts of familial and sporadic IgA nephropathy may have increased the risk of genetic kidney disease, which occurs in up to 20% of those with kidney failure (52). Even AD Alport syndrome, where kidney function is typically normal, is associated with worse function in IgA nephropathy (11, 12).
IgA vasculitis was studied here because its mesangial IgA deposits resemble those found in IgA nephropathy, and its childhood onset suggested a genetic basis. However, no coexisting genetic kidney disease was found with IgA vasculitis, and an association with HLA type has been better substantiated (22).
The strengths of this study were the unbiased cohort with familial IgA nephropathy; the additional sequencing of members of most families; examination of a cohort with sporadic biopsy-proven IgA nephropathy; investigation of a large panel of genes associated with genetic kidney disease; and the use of rigorous filtering criteria and confirmation of pathogenicity in Alamut and ClinVar.
This study’s limitations were that many index cases had kidney failure, where genetic disease is more common anyway (52); clinical information, apart from hematuria, proteinuria and kidney failure, was limited and follow up was often incomplete; variants themselves were not confirmed independently with an alternate sequencing method; the original kidney biopsies were not available for further analysis, and in most cases the diagnosis of IgA nephropathy was made on a biopsy in one family member and presumed in others with kidney abnormalities. Further, pathogenic variants might have been overlooked because the regions of some genes were not adequately covered by WES.
In conclusion, many, and possibly most, familial IgA nephropathy results from IgA deposits and coexisting genetic kidney disease, especially the more common AD and XL Alport syndrome. Sometimes, genetic kidney disease is not detected in the proband but is present in another family member with IgA nephropathy. Genetic kidney disease also occurs in sporadic IgA nephropathy with kidney failure. Individuals with familial IgA nephropathy should be offered genetic testing, and family members should only be used as kidney donors if a genetic kidney disease has been excluded. Further, more sensitive testing may demonstrate that all cases of familial IgA nephropathy result from co-existing genetic kidney disease.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1D'Amico G. The commonest glomerulonephritis in the world: Ig A nephropathy. Q J Med. (1987) 64:709–27.3329736 · pubmed ↗
- 2Schena FP Nistor I. Epidemiology of Ig A nephropathy: a global perspective. Semin Nephrol. (2018) 38:435–42. doi: 10.1016/j.semnephrol.2018.05.013, 30177015 · doi ↗ · pubmed ↗
- 3D'Amico G. Natural history of idiopathic Ig A nephropathy: role of clinical and histological prognostic factors. Am J Kidney Dis. (2000) 36:227–37. doi: 10.1053/ajkd.2000.896610922300 · doi ↗ · pubmed ↗
- 4Zhang L Liu X Pascoe EM Badve SV Boudville NC Clayton PA . Long-term outcomes of end-stage kidney disease for patients with Ig A nephropathy: a multi-centre registry study. Nephrology. (2016) 21:387–96. doi: 10.1111/nep.12629, 26393772 · doi ↗ · pubmed ↗
- 5Suzuki H Kiryluk K Novak J Moldoveanu Z Herr AB Renfrow MB . The pathophysiology of Ig A nephropathy. J Am Soc Nephrol. (2011) 22:1795–803. doi: 10.1681/ASN.2011050464, 21949093 PMC 3892742 · doi ↗ · pubmed ↗
- 6Kim CJ Woo YJ Kook H Choi YY Ma JS Hwang TJ. Henoch-Schonlein purpura nephritis associated with Epstein-Barr virus infection in twins. Pediatr Nephrol. (2004) 19:247–8. doi: 10.1007/s 00467-003-1387-7, 14677057 · doi ↗ · pubmed ↗
- 7Hiki Y Odani H Takahashi M Yasuda Y Nishimoto A Iwase H . Mass spectrometry proves under-O-glycosylation of glomerular Ig A 1 in Ig A nephropathy. Kidney Int. (2001) 59:1077–85. doi: 10.1046/j.1523-1755.2001.0590031077.x, 11231363 · doi ↗ · pubmed ↗
- 8Tomana M Novak J Julian BA Matousovic K Konecny K Mestecky J. Circulating immune complexes in Ig A nephropathy consist of Ig A 1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. (1999) 104:73–81. doi: 10.1172/JCI 5535, 10393701 PMC 408399 · doi ↗ · pubmed ↗