Do Not Dismiss Incidental Hyperkalemia in Childhood: Early Recognition of Pseudohypoaldosteronism Type II
Hiroaki Kanai, Hiroki Sato

TL;DR
A child's persistent hyperkalemia led to the early diagnosis of pseudohypoaldosteronism type II, highlighting the importance of thorough evaluation in children.
Contribution
The paper emphasizes the importance of not dismissing incidental hyperkalemia in children and provides a structured approach for early diagnosis of PHAII.
Findings
Persistent hyperkalemia in a child was linked to pseudohypoaldosteronism type II (PHAII).
A KLHL3 gene variant was identified through genetic testing, confirming the PHAII diagnosis.
Treatment with hydrochlorothiazide normalized potassium levels and corrected metabolic acidosis.
Abstract
Persistent hyperkalemia in children warrants careful evaluation, as it may indicate an underlying renal tubular disorder. We report a case of persistent hyperkalemia in a three-year-old boy in which adenovirus infection unmasked latent pseudohypoaldosteronism type II (PHAII), allowing recognition prior to the development of hypertension. The patient presented with a nine-day history of fever. Initial laboratory tests showed hyperkalemia (5.8 mmol/L), mild hyponatremia, normal renal function, and normal anion gap (AG) metabolic acidosis (pH: 7.37; bicarbonate: 19.9 mmol/L; AG: 8.1 mmol/L). The patient was diagnosed with adenovirus infection. Although the fever and inflammatory markers improved within four days, the hyperkalemia persisted (6.2 mmol/L). At three and six weeks, serum potassium remained elevated (6.0 and 6.4 mmol/L, respectively) with normal AG metabolic acidosis (HCO₃⁻ 20.0…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
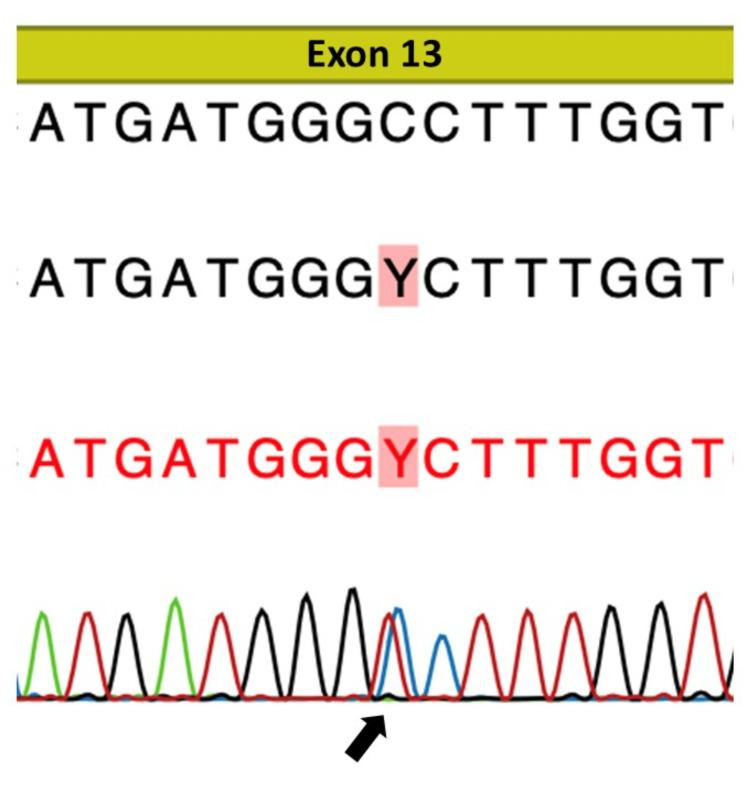 Figure 1
Figure 1| Variable | On presentation | Four days after presentation | Three weeks after presentation | Six weeks after presentationa | One month after HCTZ initiation | Three months after HTCZ initiationb | One month after HCTZ dose escalation | Reference value |
| WBC count (× 103 cells/µL) | 14.7 | 11.9 | 9.9 | 8.6 | - | - | - | 3.9-9.8 |
| CRP (mg/dL) | 5.99 | 1.62 | 0.34 | 0.08 | - | - | - | <0.15 |
| Creatinine (mg/dL) | 0.25 | 0.30 | 0.34 | - | - | - | - | 0.21-0.37 |
| Na (mmol/L) | 132.7 | 137.0 | 137.0 | 137.1 | 139.5 | 139.5 | 136.9 | 138-145 |
| K (mmol/L) | 5.8 | 6.2 | 6.0 | 6.4 | 4.7 | 5.8 | 4.6 | 3.6-4.8 |
| Cl (mmol/L) | 104 | 105 | 108 | 109 | 105 | 110 | 105 | 101-108 |
| pH | 7.37 | n.a. | 7.31 | 7.28 | 7.36 | 7.33 | 7.38 | 7.32-7.41 |
| PCO2 (mmHg) | 34.9 | n.a. | 40.9 | 38.1 | 46.2 | 37.8 | 42.3 | 42-53 |
| HCO₃⁻ (mmol/L) | 19.9 | n.a. | 20.0 | 17.6 | 25.2 | 19.6 | 24.6 | 24-28 |
| AG (mmol/L) | 8.1 | n.a. | 12.8 | 11.4 | 12.4 | 11.4 | 10.3 | 7-13 |
| Plasma renin activity (ng/mL/h) | n.a. | n.a. | n.a. | 0.6 | - | - | - | 0.2-2.3 |
| Aldosterone (pg/mL) | n.a. | n.a. | n.a. | 38.0 | - | - | - | 29.9-158.8 |
| Variable | Three weeks after presentation | Six weeks after presentation | Reference value |
| FEK (%) | 7.6 | 9.1 | >20% during hyperkalemia |
| TTKGa | 2.9 | 5.3 | >6 during hyperkalemia |
| Urinary Na (mmol/L) | 146.3 | 212.9 | n.a. |
| Urine osmolality (mOsm/kg) | 1181 | 921 | 200-800 |
| Plasma osmolality (mOsm/kg) | 286 | 291 | 275-285 |
| Reference | Age | Reason for the blood tests | K (mmol/L) | HCO3- (mmol/L) | Hypertension | Mutated gene |
| Tsuji et al., 2013 [ | 3 years | Croup | 6.8 | 12.6 | + | CUL3 |
| Mitani et al., 2016 [ | 10 months | Loss of consciousness | 6.6 | 14.8 | + | KLHL3 |
| Hollander et al., 2016 [ | 6 years | Hematuria | 8.6 | 16 | + | CUL3 |
| Hollander et al., 2016 [ | 4 months | Decrease in enteral feeding | 7.0 | 22 | + | KLHL3 |
| Park et al., 2017 [ | 9 months | Urinary tract infection | 6.3 | 13 | + | KLHL3 |
| Park et al., 2017 [ | 20 months | Intussusception | 6.9 | 12 | + | KLHL3 |
| Doan et al., 2020 [ | 2 months | Feeding difficulty | 6.6 | n.a. | n.a. | KLHL3 |
| Yavas Abali et al., 2020 [ | 14 years | Short stature, excessive weight gain | 6.4 | n.a. | + | CUL3 |
| Nakano et al., 2020 [ | 2 months | Respiratory syncytial virus infection | n.a. | n.a. | n.a. | CUL3 |
| Ostrosky-Frid et al., 2020 [ | 12 years | Hypertension | 7.0 | 17.8 | + | CUL3 |
| Patti et al., 2021 [ | 19 months | Fever | 7.45 | 15.2 | + | CUL3 |
| Park et al., 2022 [ | 7 years | Short stature | 6.8 | 16.7 | + | CUL3 |
| Babar et al., 2022 [ | 5 weeks | Difficulty in feeding, lethargy, emesis | 10.1 | n.a. | − | WNK1 |
| Zieg et al., 2025 [ | 3 years | Preoperative work-up | 7.6 | 11.3 | + | KLHL3 |
| Zieg et al., 2025 [ | 10 days | Insufficient weight gain | 7.4 | 16 | n.a. | KLHL3 |
| Modi et al., 2025 [ | 14 years | Abdominal pain, vomiting, watery diarrhea | 5.6 | 19 | − | WNK1 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIon Transport and Channel Regulation · Vitamin K Research Studies · Helicobacter pylori-related gastroenterology studies
Introduction
Hyperkalemia can result from reduced renal potassium excretion, excessive potassium intake, medication effects, or a transcellular shift due to acidosis, diabetes mellitus, or massive tissue breakdown, as seen in rhabdomyolysis [1,2]. Spurious hyperkalemia is common in pediatric practice, often due to hemolysis during blood sampling; however, true hyperkalemia in children is rare [3]. Consequently, incidental findings are often dismissed as transient or artifactual [2]. However, persistent hyperkalemia warrants systematic evaluation, as it may indicate an underlying renal tubular disorder.
Pseudohypoaldosteronism type II (PHAII) is a monogenic disorder characterized by hypertension, hyperkalemia, and metabolic acidosis with a normal anion gap (AG) [3,4]. It involves sodium-chloride cotransporter (NCC) activity in the distal convoluted tubule [4]. NCC overactivity enhances sodium and chloride reabsorption [5]. The consequent impaired delivery of sodium to the collecting duct results in impaired aldosterone-mediated exchange of sodium for potassium and protons, resulting in hyperkalemia and hyperchloremic metabolic acidosis, despite normal aldosterone production [5]. This disease is considered to be rare; however, the true incidence is unknown because the available evidence is limited to case reports and small case series, with no population-based studies. It is likely underdiagnosed, not only in children but also in adults with hypertension, as genetic testing is not routinely performed. Although hypertension usually develops in late childhood or even adulthood, the metabolic disorders, hyperkalemia and metabolic acidosis, can precede hypertension and may be present at birth [5,6]. Therefore, PHAII should be considered in the differential diagnosis of unexplained hyperkalemia in children of all ages, even if they are normotensive. Here, we report a pediatric case of hyperkalemia in which adenovirus infection unmasked previously latent PHAII, allowing recognition of the disease in an early stage prior to the onset of hypertension.
Case presentation
A previously healthy three-year-old boy with normal growth and development presented with a nine-day history of fever. Initial laboratory test results revealed hyperkalemia and low sodium levels, with chloride and creatinine levels within normal limits. Venous blood gas analysis was consistent with metabolic acidosis with partial respiratory compensation. The white blood cell (WBC) count and C-reactive protein (CRP) level were elevated, indicating acute inflammation (Table 1). A respiratory panel confirmed adenovirus infection.
Four days later, the fever had resolved, and the WBC count and CRP level had decreased; however, the hyperkalemia persisted. Three weeks after presentation, the patient was re-evaluated for persistent hyperkalemia. His height was 99.0 cm, weight was 16.2 kg, and blood pressure (96/55 mmHg) was normal for a child of his age. Blood tests revealed persistent hyperkalemia with normal AG metabolic acidosis (Table 1). The urinary indices suggested reduced renal potassium excretion, with a reduced fractional excretion of potassium (FEK) and transtubular potassium gradient (TTKG) (Table 2). At this time, the urinary sodium concentration was high, and the urine osmolality exceeded the plasma osmolality (Table 2), supporting a valid interpretation of TTKG [7]. Six weeks after presentation, the hyperkalemia with normal AG metabolic acidosis persisted (Table 1), with reduced FEK and TTKG (Table 2). Again, the urinary sodium concentration was high, and the urine osmolality exceeded the plasma osmolality (Table 2); however, the plasma renin activity and aldosterone levels were within normal limits (Table 1). Renal ultrasonography showed no abnormalities. At this time, the patient’s father reported a history of hypertension, having been diagnosed in his twenties.
The constellation of persistent hyperkalemia, normal AG metabolic acidosis, reduced renal potassium excretion, and a family history of early-onset hypertension suggested PHAII. Treatment with hydrochlorothiazide (HCTZ) was initiated at a dose of 0.7 mg/kg/day, and on retesting one month later, the potassium and bicarbonate levels were within the reference range. However, retesting two months later (three months after initiating HCTZ treatment) revealed a recurrence of hyperkalemia and normal AG metabolic acidosis. The dose of HCTZ was increased to 1.5 mg/kg/day, and on retesting one month later, the potassium and bicarbonate levels were again within the reference range. The serial laboratory findings during the clinical course are summarized in Table 1.
Gene sequence analysis identified a heterozygous missense mutation in KLHL3 (c.1501C>T, p.Pro501Ser, Exon 13) (Figure 1), listed in the Human Gene Mutation Database (ID: 22266938) and classified as a variant of uncertain significance. However, considering the biochemical phenotype and clear responsiveness to thiazide therapy, we judged this variant to be pathogenic.
Gene sequencing resultsThe figure shows a heterozygous C-to-T substitution at nucleotide position c.1501 in exon 13 (c.1501C>T; arrow), resulting in a heterozygous missense mutation. The change involves a substitution of proline to serine at position 501 (p.Pro501Ser)
Discussion
PHAII, also known as Gordon syndrome, is a renal tubular disorder caused by increased NCC activity in the distal convoluted tubule, leading to increased sodium reabsorption and decreased potassium and hydrogen excretion [5]. Causative mutations involve WNK1, WNK4, KLHL3, and CUL3 [6]. KLHL3 and CUL3 were first recognized as causative genes in 2012 [8]. Most cases follow an autosomal dominant inheritance pattern, although autosomal recessive KLHL3 variants have been reported [4]. Phenotypic severity varies by genotype, ranked in decreasing order as follows: CUL3, recessive KLHL3, dominant KLHL3, WNK4, and WNK1 [6,8]. PHAII responds well to treatment with thiazide diuretics, which directly inhibit NCC, correcting the electrolyte imbalance and preventing hypertension.
A PubMed search of PHAII cases in children aged under 15 years, published since 2012 (when KLHL3 and CUL3 gene variants were recognized as causative), identified case reports in which the reason for the blood tests was explicitly reported (Table 3) [3,5,9-19]. Almost all cases were detected incidentally, typically during evaluations of unrelated conditions such as acute illness, poor growth or feeding, preoperative testing, or routine laboratory screening. The PubMed search identified only one child with PHAII who was diagnosed specifically as a result of investigating unexplained hypertension. However, despite PHAII being discovered incidentally, most children in published pediatric case reports already exhibited hypertension. KLHL3 and CUL3 variants predominated among the reported cases, reflecting the earlier and more severe phenotypes associated with variants in these genes. Our patient presented with fever due to an adenovirus infection, initially raising the question of whether acute illness or dehydration contributed to the electrolyte abnormalities. However, the persistent hyperkalemia and metabolic acidosis after complete recovery indicate that the infection was not the primary cause. Instead, the acute illness prompted the laboratory testing that led to the diagnosis. This case differs from most previously reported cases in that hypertension was absent, representing an early biochemical stage of PHAII and providing insight into early disease evolution.
In this case, persistent hyperkalemia despite preserved glomerular function, accompanied by normal AG metabolic acidosis, led us to conduct further investigations. These revealed impaired renal potassium excretion (low FEK and TTKG values), which led us to suspect PHAII. This case offers several important educational points. First, incidental hyperkalemia in children should not be dismissed without confirmation. Although pseudohyperkalemia is common in children, clinicians should not attribute it solely to transient or artifactual causes, and repeated testing to verify persistent hyperkalemia is a crucial first step to diagnosis, even in mild or asymptomatic hyperkalemia. In addition, clinicians should also assess the acid-base balance carefully because concomitant normal AG metabolic acidosis is an important clue to diagnosis and suggests a renal tubular disorder rather than a systemic cause. Second, urinary potassium excretion should be assessed. Urinary potassium indices are simple, but essential diagnostic markers. Persistently low FEK and TTKG values during hyperkalemia indicate impaired renal tubular excretion [1]. The FEK values provide evidence of impaired renal potassium handling. Additionally, the TTKG, which reflects aldosterone-dependent potassium secretion in the distal nephron, is particularly informative when interpreted under appropriate physiological circumstances, specifically, urinary sodium concentration >25 mmol/L and urine osmolality greater than plasma osmolality [7]. In this case, persistently low FEK and TTKG values during hyperkalemia, combined with urinary sodium concentrations >25 mmol/L and urine osmolality greater than plasma osmolality at both urinary assessments, provided strong evidence of inappropriate low urinary potassium secretion, despite persistent hyperkalemia. Finally, given the autosomal dominant inheritance pattern in most PHAII cases, a family history of early-onset hypertension is an important diagnostic clue, even when the child is normotensive [8]. In this case, the father’s early-onset hypertension, diagnosed in his twenties, was only identified at the patient’s fourth visit, suggesting that earlier inquiry into family history might have expedited diagnosis. This highlights the importance of obtaining a detailed family history during the initial evaluation of persistent hyperkalemia. Greater awareness of these principles may facilitate earlier detection of PHAII, enabling timely treatment and preventing progression to hypertension and its complications.
This report has some limitations. It describes a single case, so the generalizability of our findings may be limited. In addition, the identified KLHL3 variant is classified as a variant of uncertain significance, although the clinical phenotype and therapeutic response to thiazide diuretics in this case strongly support its pathogenicity.
Conclusions
This case highlights the importance of not dismissing incidentally detected hyperkalemia in children and illustrates the value of using a structured, stepwise approach to the investigation and management of pediatric hyperkalemia, and of confirming its persistence through repeated measurement. If the hyperkalemia persists, acid-base balance (pH and bicarbonate level) and renal potassium excretion should be assessed, and inquiries should be made about a possible family history of early-onset hypertension. This structured approach can identify renal tubular disorders such as PHAII even in apparently healthy children, preventing diagnostic delays and ensuring timely, effective management.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pathogenesis, diagnosis and management of hyperkalemia Pediatr Nephrol Lehnhardt A Kemper MJ 3773842620112118120810.1007/s 00467-010-1699-3PMC 3061004 · doi ↗ · pubmed ↗
- 2Potassium Nelson Textbook of Pediatrics. 22nd edition Greenbaum LA 495501 Elsevier 12024 https://shop.elsevier.com/books/nelson-textbook-of-pediatrics-2-volume-set/kliegman/978-0-323-88305-4
- 3Hyperkalemia in young children: blood pressure checked?Eur J Pediatr Hollander R Mortier G van Hoeck K 2011201317520162763985710.1007/s 00431-016-2782-y · doi ↗ · pubmed ↗
- 4Gordon syndrome: a continuing story Pediatr Nephrol O'Shaughnessy KM 190319083020152550332310.1007/s 00467-014-2956-7 · doi ↗ · pubmed ↗
- 5Hyperkalaemic acidosis: blood pressure is the diagnostic clue Pediatr Nephrol Zieg J ThomasováD Libik M Sumnik Z Bockenhauer D 967970402025 https://pubmed.ncbi.nlm.nih.gov/39527282/3952728210.1007/s 00467-024-06590-4PMC 11885314 · doi ↗ · pubmed ↗
- 6The molecular genetics of Gordon syndrome Genes (Basel) Mabillard H Sayer JA 9861020193179549110.3390/genes 10120986 PMC 6947027 · doi ↗ · pubmed ↗
- 7Normal reference values Pediatric Nephrology. Eighth ed Cano F Gajardo M Shen Q Nehus E Dixon B 20492078 Cham Springer 2022 https://link.springer.com/rwe/10.1007/978-3-030-52719-8_142
- 8Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities Nature Boyden LM Choi M Choate KA 9810248220122226693810.1038/nature 10814 PMC 3278668 · doi ↗ · pubmed ↗