CLL-1: An emerging target for immunotherapy in acute myeloid leukemia
Yu Wang, Yi Xiao

TL;DR
This paper reviews CLL-1 as a new target for immunotherapy in acute myeloid leukemia, offering potential for more effective and less toxic treatments.
Contribution
The paper highlights CLL-1 as a novel immunotherapy target in AML with specific expression in leukemia cells.
Findings
CLL-1 is specifically expressed in AML blast cells and leukemia stem cells.
Targeting CLL-1 may reduce drug resistance and non-specific toxicity.
CLL-1-targeted therapies could help overcome immune escape in AML.
Abstract
AML is an aggressive haematological malignancy characterised by uncontrolled proliferation and differentiation arrest of myeloid progenitor/stem cells. Conventional treatment methods principally entail chemotherapy and haematopoietic stem cell transplantation; however, the efficacy of these treatments is constrained by the occurrence of relapses and treatment-related toxicity. In recent years, research into molecular mechanisms has driven the development of targeted therapies against specific gene mutations and advanced multiple immunotherapy strategies. Among these, C-type lectin-like molecule 1 (CLL-1) has emerged as a promising new immunotherapy target due to its specific expression in AML blast cells and leukemia stem cells. CLL-1-targeted therapies have been shown to have the potential to alleviate drug resistance, reduce non-specific toxicity, and address issues of immune escape.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
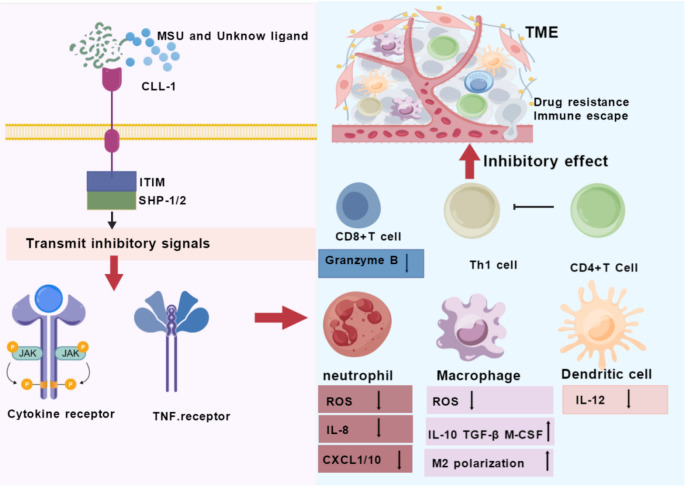 Figure 1
Figure 1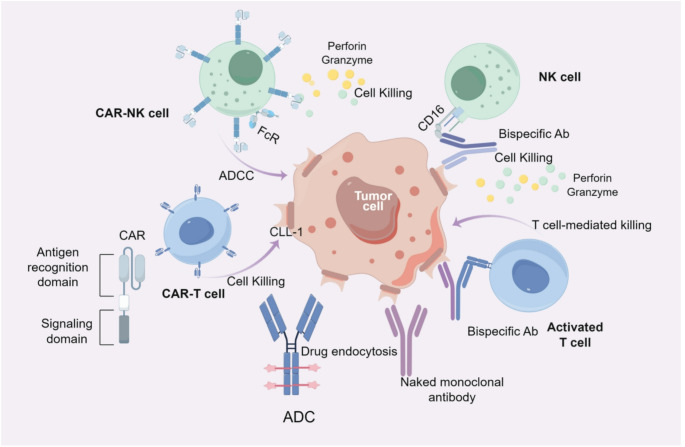 Figure 2
Figure 2- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · Chronic Lymphocytic Leukemia Research · Phagocytosis and Immune Regulation
Introduction
AML is one of the most common types of acute leukaemia in adults. It is characterised by high heterogeneity and aggressiveness. Its pathogenesis primarily involves the abnormal proliferation and impaired differentiation of myeloid progenitor cells [1]. Traditional treatment paradigms for AML mainly rely on chemotherapy and haematopoietic stem cell transplantation [2], however, the efficacy of chemotherapy is often limited by relapse, drug resistance and treatment-related toxicity, while transplantation is restricted by donor availability, transplant-related complications and graft-versus-host disease [3, 4]. With the rapid development of molecular pathology and genomics research, targeted drugs for specific gene abnormalities have gradually entered clinical use. Examples include the FLT3, IDH1 and IDH2 inhibitors, which have improved the prognosis of some patients [5–7]. However, due to the complex molecular spectrum and significant heterogeneity of AML, monotherapy with targeted drugs alone is insufficient to comprehensively address relapse and drug resistance. Therefore, immunotherapy for AML has recently become a popular research topic, with various strategies being investigated, including monoclonal antibodies, bispecific antibodies, and Chimeric Antigen Receptor T-cell (CAR-T) cell therapy [8–10]. Of particular interest is the CLL-1, which is highly expressed in AML blasts and leukaemia stem cells, but absent or only weakly expressed in normal haematopoietic stem cells [11–13]. This demonstrates good target specificity and safety. Research into immunotherapy centred on CLL-1 offers new ideas for overcoming the challenges of drug resistance, toxicity and immune evasion in current treatments. This article provides a systematic review of CLL-1-related research and explores its potential value in AML treatment. The aim is to promote the optimisation of precision treatment strategies and the clinical translation of novel immunotherapies.
Biological characteristics of CLL-1
C-type lectin-like molecules (CTLs) are a large and highly diverse superfamily of proteins characterised by a carbohydrate-recognition domain (CRD) which typically relies on calcium ions (Ca²⁺) to bind specifically to certain glycan ligands [14]. These molecules are widely expressed on the surface of immune cells and play an indispensable role in biological processes such as immune recognition, cell adhesion, inflammation regulation and host defence mechanisms against pathogens [15, 16]. CLL-1, also known as CLEC12A) is an important member of this family. The CLL-1 gene is located in the p13.31-p13.2 region of human chromosome 12 and encodes 265 amino acids [11]. Structurally, CLL-1 exhibits the typical features of a type II transmembrane protein, with its extracellular segment containing a highly conserved C-type lectin-like domain [17, 18]. While CLL-1 is categorised as part of the C-type lectin family, it is important to note that its CRD domain has undergone significant amino acid substitutions, replacing the conserved ‘EPN’ motif with a ‘QPD’ motif. This results in the loss of the ability to bind to classical Ca²⁺-dependent glycans and instead confer specificity to certain endogenous ligands, such as urate crystals released during cell death [19]. This unique structural feature determines its functional orientation. At a molecular level, CLL-1 primarily transmits inhibitory signals through its intracellular segment via the immunoreceptor tyrosine-based inhibitory motif (ITIM). Upon ligand binding, CLL-1 negatively regulates cell signalling pathways mediated by activating receptors, thus acting as a ‘brake’ to maintain immune homeostasis and prevent autoimmune reactions [20]. In particular, on myeloid leukemia cells, CLL-1, as a specific surface antigen, is closely related to the survival and proliferation of tumor cells, placing CLL-1 at the core of immune surveillance. Therefore, CLL-1 holds significant value as an emerging target for AML immunotherapy. The structural differences between CLL-1 and other AML-related targets are shown in Table 1.
Table 1. Structural differences of common targets in AMLTargetStructural Features Signaling PathwayLigand-Binding PropertiesPropertiesExpression Pattern in AMLExpression in Healthy TissuesClinical Stage of Targeted TherapyCLL-1Single-pass transmembrane, CRD domainSyk/Src-PI3K/AKTGlycosylated proteins> 90% AML blasts; high on LSCs; variable across FAB (M4/M5 high, M0/M3 low) [11, 13]Myeloid lineage (neutrophils, monocytes, macrophages, DCs); absent on HSCs [13]Phase I/II (CAR-T: NCT07036250; BsAb: NCT03038230; ADC: preclinical)CD123Three-pass transmembrane, IL-3 receptor α-chainJAK/STATIL-380–93% AML; IL−3Rα chain; high on LSCs [21, 22]Endothelial cells, plasmacytoid DCs, basophils; low on HSCs [21]Phase I/II (BsAb: flotetuzumab NCT02152956;CAR-T: NCT04318678ADC: tagraxofusp NCT02268253)CD33Single-pass transmembrane, Ig-like domainITIM-mediated phosphatase recruitmentSialic acid derivatives85–90% AML; expressed on LSCs [22]Myeloid progenitors, mature myeloid cells; low on HSCs [23]Approved (GO, 2017 reapproval); Phase I/II (CAR-T: NCT06420063;BsAb: NCT05077423)CD47Five-pass transmembrane, Ig superfamily ligandBinds SIRPα, transmits “don’t eat me” signal, inhibits macrophage phagocytosisMacrophage surface SIRPα proteinUniversal expression on AML; “don’t eat me” signal [24]Ubiquitous (all normal cells); high on RBCs, platelets [25]Phase I/II (Magrolimab; BsAb: evorpacept)TIM-3Single-pass transmembrane, Ig V-like domainNo classic intracellular motif; delivers inhibitory signals via co-receptors (negative T/NK regulation)Galactin-9, phosphatidylserine, etc.80–90% AML; co-expressed with LSC markers; immune checkpoint [26, 27]T cells, NK cells, monocytes (immune cells); not tumor-specific [28]Phase I/II (Sabatolimab + HMAs); Phase III ongoing (STELLAR−001)AML Acute Myeloid Leukemia, IL Interleukin, DC Dendritic Cell, HSCs Hematopoietic Stem Cells, LSCs Leukemic Stem Cells, GO Gemtuzumab ozogamicin, RBCs Red Blood Cells, NK Natural Killer (Cell), HMA Hypomethylating Agent
Expression and clinical significance of CLL−1
Tissue distribution and phylogenetic restriction
The expression profile of CLL-1 (CLEC12A) is characterised by significant tissue- and lineage-restriction, with its expression being predominantly confined to the hematopoietic system, particularly within myeloid cells. The CLL-1 protein has been detected on the surface of a range of terminally differentiated myeloid immune cells in peripheral blood and bone marrow. These cells include neutrophils, monocytes, macrophages, and certain subsets of dendritic cells [11]. Conversely, no substantial expression of CLL-1 has been observed in lymphoid cells, including T lymphocytes and B lymphocytes [29], highlighting its potential value as a marker of myeloid differentiation.
Expression patterns in hematologic malignancies
The expression profile of CLL-1 is of significant clinical significance in the context of haematological malignancies. In AML, CLL-1 is persistently overexpressed on tumor cells. Research data indicate that the transmembrane form of CLL-1 glycoprotein is detectable in over 90% of AML cases [29]. Multiparameter flow cytometry analysis reveals that CLL-1 is present across all French-American-British (FAB) subtypes, albeit with divergent expression intensities that correlate with the degree of monocytic differentiation. Specifically, the highest mean positivity rates and fluorescence intensities are observed in M4 (myelomonocytic) and M5 (monocytic) subtypes, which are characterized by prominent monocytic/granulomonocytic differentiation. This elevated expression aligns with the physiological abundance of CLL-1 on normal monocytes and macrophages, suggesting that leukemic cells in these subtypes retain differentiation antigen expression patterns consistent with their monocytic lineage commitment. Conversely, M3 (acute promyelocytic leukemia, APL) demonstrates the lowest CLL-1 expression levels, likely reflecting its distinct pathogenesis driven by the PML-RARA fusion and its early myeloid progenitor origin with minimal monocytic differentiation potential. Notably, M0 (minimally differentiated AML) exhibits the weakest CLL-1 expression overall, consistent with its primitive immunophenotype characterized by limited myeloid maturation markers [11, 30]. Importantly, this subtype-specific expression pattern suggests that CLL−1 may serve not merely as a leukemia-associated antigen, but as a marker reflecting the differentiation stage and lineage fidelity of the malignant clone. Furthermore, no substantial disparities in CLL-1 expression levels have been identified among specific molecular genetic subtypes, including TP53 and NPM1 mutations [31], indicating that CLL-1 overexpression in AML is relatively independent of these common molecular alterations.
Beyond AML, CLL-1 expression demonstrates significant heterogeneity in myelodysplastic syndromes (MDS) and chronic myeloid leukemia (CML). Combined flow cytometry and RNA-sequencing analyses reveal that the positivity rate of CLL-1 on CD34 + progenitor cells in MDS patients fluctuates between 10% and 45%. In CML, CD34 + CD38- progenitor cell subsets in the chronic phase exhibit minimal to no CLL-1 expression; however, during accelerated or blast crisis phases, positivity rates surge to 25%–60%, suggesting a close correlation with leukemic transformation [18, 29]. This dynamic regulation—ranging from absent/low expression in normal early progenitors and chronic-phase CML to marked upregulation in differentiated myeloid malignancies and blast crisis—reinforces the association between CLL-1 expression and myeloid maturation. Consequently, therapeutic strategies targeting this antigen may specifically eliminate leukemia cells while minimizing impact on normal hematopoietic stem cells that lack CLL-1 expression. As research advances, therapeutic interventions targeting CLL-1 are anticipated to expand beyond AML to encompass other hematological malignancies, including high-risk MDS and blast-phase CML.
Prognostic significance
Prognostically, CLL-1 expression levels demonstrate significant clinical correlation with patient outcomes. A study of 123 CD34 + AML patients revealed that low CLL-1 expression (CLL-1 low) was significantly associated with reduced complete remission(CR) rates (OR = 4.57) and elevated minimal residual disease(MRD) levels following induction chemotherapy [31]. Multivariate analysis confirmed that CLL-1 low status represents an independent adverse prognostic factor for both event-free survival and overall survival [31], with predictive value that transcends traditional risk stratification systems. Interestingly, this prognostic association appears somewhat paradoxical given the higher CLL−1 expression observed in more differentiated FAB subtypes (M4/M5), which are themselves associated with distinct clinical features. This suggests that within the context of CD34 + AML—a population typically enriched for less differentiated disease—retained CLL−1 expression may identify a subgroup with relatively more mature differentiation characteristics and correspondingly better treatment response. This prognostic utility aids in identifying patients with poor prognosis within intermediate and low-risk cytogenetic categories, potentially guiding therapeutic intensification strategies.
Challenges and strategies for therapeutic translation
The specific expression of CLL-1 in myeloid tumors makes it a highly promising therapeutic target, and it has demonstrated preliminary efficacy in clinical studies [32, 33]. However, CLL-1-targeted therapy also carries risks such as bone marrow suppression and infection. simultaneously, the expression heterogeneity of CLL-1 across FAB subtypes, particularly its significantly reduced expression in M0 (differentiated) and M3 (acute promyelocytic leukemia) subtypes, poses substantial challenges to monotherapy strategies. Single-antigen targeting may prove insufficient for complete eradication of malignant clones. Therefore, to further enhance the efficacy of CLL-1-targeted therapies and overcome antigen escape induced by single-target approaches, multi-target combination strategies are emerging as a key direction for preventing antigen escape-mediated relapse. Currently, bispecific antibodies targeting CLL-1 (e.g., CLL-1/CD3 BsAbs) and composite CAR-T cells (e.g., CLL-1/CD33 cCAR-T) have both demonstrated encouraging clinical efficacy [34, 35]. Additionally, combination therapies like CLL-1/CD123 tandem CAR-T cells have demonstrated enhanced cytotoxicity against CD123+/CLL-1 + leukemia cells [36]. These multi-target strategies effectively address treatment challenges posed by FAB classification heterogeneity by simultaneously targeting non-overlapping antigen expression profiles, providing a crucial pathway to overcome resistance mechanisms mediated by single-antigen loss mutations.
CLL-1-Mediated signaling pathways
CLL-1, a member of the C-type lectin-like receptor family, contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) in its cytoplasmic tail. It can thus be concluded that the core characteristic of the signal transduction mechanism under investigation is the transmission of inhibitory signals as opposed to activating signals [11]. Upon binding of CLL-1 to its ligands, such as sodium urate crystals or as-yet-unidentified cell surface molecules, Src family kinases are rapidly phosphorylated and recruit SHP-1/2 phosphatases, forming a myeloid-specific negative regulatory node. This node dephosphorylates neighbouring Syk, TRAF6, or IKKβ, thereby interrupting the TLR4-NF-κB and FcγR-ROS cascades [29]. The resultant effect of this process is a decrease in IL-12p70, an increase in IL-10, and the inhibition of neutrophil calcium flux and superoxide release. In summary, in AML, the binding of CLL-1 to its ligands weakens the innate immune clearance of leukemia cells by the body. Concurrently, the downregulation of IL-12 and TNF-α, concomitant with the upregulation of IL-10, further induces the bone marrow microenvironment into an immunosuppressive state, thereby engendering conditions conducive to chemotherapy resistance.
CLL-1 and immune cells
Upon binding to its ligands, CLL-1 transmits inhibitory signals to multiple immune cells via the ITIM-SHP-1/2 axis [37], as demonstrated in Fig. 1. In the case of neutrophils, this pathway has been shown to rapidly block the binding of the NADPH oxidase complex within minutes, thereby inhibiting the production of superoxide anions (ROS) and IL-8, and downregulating the expression of chemokines such as CXC chemokine ligand 1 (CXCL1) and CXC chemokine ligand 10 (CXCL10). This results in the limitation of the recruitment and infiltration of neutrophils at sites of inflammation, whilst concurrently reducing calcium flux and degranulation, thus giving rise to a hyporesponsive or “quiescent” phenotype [11, 38]. In monocytes and macrophages, SHP-1/2-mediated dephosphorylation similarly inhibits NOX2 activity, leading to reduced levels of extracellular and mitochondrial ROS [39, 40]. Reactive oxygen species (ROS) are pivotal molecules for macrophages in terms of executing oxidative damage and immune effects. It has been demonstrated through a range of studies that when the levels of ROS produced by macrophages are reduced by antioxidants or chenodeoxycholic acid (CDCA), the lipid peroxidation signal in the co-culture system with AML is also reduced. This has the effect of decreasing the apoptosis rate of the leukaemia cells and increasing their proliferation [41], The mechanism of this process is thought to lie in the disruption of the ROS-p38 MAPK-DGAT1 axis, reduced lipid droplet accumulation, and blocked ferroptosis, leading to decreased sensitivity to chemotherapy or targeted drugs. Furthermore, a low-ROS environment has been shown to drive macrophages to polarise towards the M2 type, accompanied by increased secretion of IL-10, TGF-β, and M-CSF [42], which in turn inhibits Th1 responses and promotes the self-renewal of leukemia stem cells [43, 44]. This inhibitory signal has been demonstrated to downregulate the levels of CD80/CD86 in dendritic cells and to reduce IL-12 secretion. Consequently, it has been shown to inhibit the polarization of CD4 + T cells towards Th1 and to downregulate the expression of granzyme B in CD8 + T cells. This, in turn, has been shown to weaken the cytotoxic T lymphocyte (CTL) response [29]. Consequently, despite the absence of CLL-1 expression on the surface of NK/T cells, it is capable of exerting an indirect inhibitory effect on the adaptive immune effector functions by means of altering the cytokine network.
Fig. 1CLL−1-mediated immunoinhibitory signaling in the AML microenvironment. Binding of MSU or unknown ligands to CLL-1 recruits SHP-1/2 via the ITIM motif, inhibiting downstream cytokine and TNF receptor signaling. In the TME, this suppresses neutrophil oxidative burst and chemotaxis, promotes macrophage M2 polarization, reduces dendritic cell IL-12 production, and impairs Th1 differentiation and CD8 + T cell cytotoxicity, driving immune escape and drug resistance
CLL-1 endocytosis kinetics
The endocytic kinetics of CLL-1 are pivotal in determining its potential for development in antibody-drug conjugate (ADC) and CAR-T cell therapies. Research has demonstrated that the cytoplasmic tail of CLL-1 contains a canonical endocytic signal, thereby enabling rapid internalisation via clathrin-mediated endocytosis (CME). In experimental conditions, significant intracellular vesicle localization can be observed within a timespan of 15–30 min. Fluorescence pulse-chase experiments indicate that over 70% of CLL-1 molecules enter early endosomes within one hour, followed by sorting to late endosomes/lysosomes, with a degradation rate higher than the recycling rate [45]. This characteristic is advantageous for ADCs, as the antibody-bound drug can rapidly internalize and accumulate in lysosomes, efficiently releasing the payload. Preclinical models have confirmed that CLL-1-ADCs (with PBD or DM4) achieve an IC50 of less than 50 pmol·L⁻¹ against primary AML cells, with no target mutations observed in resistant strains [46]. This finding indicates that the antigen does not evade elimination through endocytic depletion.
In the context of CAR-T therapies, the endocytic kinetics have been demonstrated to exert a significant influence on the signal persistence and exhaustion profiles. The rapid internalisation of CLL-1 has been demonstrated to significantly reduce target antigen levels in the immunological synapse within a short period, which in turn leads to the indirect attenuation of the first signal. Whilst this process assists in the reduction of tonic stimulation and the delay of T cell exhaustion, excessive downregulation has also been demonstrated to induce instability within the synapse and to diminish continuous killing. Experiments have demonstrated that when the binding affinity of the CAR domain is reduced to 1–5 µmol·L-1 and a 4-1BB co-stimulatory domain is employed, phosphorylation signals can be sustained post-endocytosis, thereby achieving a balance between the efficacy of killing and the longevity of T cells [47]. It is important to note that high levels of reactive oxygen species (ROS) in the AML microenvironment have been shown to inhibit endosome acidification [48], thereby slowing the delivery of CLL-1 to lysosomes. This results in decreased ADC payload release efficiency and prolonged CAR signalling. Concurrent administration of NADPH oxidase inhibitors has been demonstrated to restore lysosomal acidity and reverse resistance. In summary, the “moderately rapid, degradation-dominant” endocytic kinetics of CLL-1 provide ADCs with the advantage of high lysosomal payload accumulation and create a low tonic stimulation environment for CAR-T therapies.
CLL-1 targeted therapy strategies
In light of the distribution characteristics and specificity of CLL-1 in AML, researchers are exploring novel immunotherapies targeting CLL-1. These include naked monoclonal antibodies, CLL-1-directed CAR-T cells, CLL-1 CAR-NK cell therapy, as well as ADCs, bispecific antibodies and trispecific antibodies based on the CLL-1 target (see Fig. 2).
Fig. 2. Schematic overview of CLL-1-directed immunotherapy modalities: naked monoclonal antibodies, internalizing ADCs, bispecific immune cell engagers, and adoptive CAR-T/NK cellular therapies
Naked monoclonal antibody
In consideration of the distribution characteristics and specificity of CLL-1 in AML, researchers have sought to explore the potential application of CLL-1 monoclonal antibodies in the treatment of AML. In preclinical experiments, it has been confirmed that unconjugated anti-CLL-1 antibodies relying solely on natural immune effects can exert strong anti-leukemia effects [49]. The murine anti-CLL-1 naked monoclonal antibody (clone 14C2) has been demonstrated to induce complement-mediated toxicity in 16 primary AML samples of various genetic subtypes, with 94% of the samples exhibiting dose-dependent complement activation and lysis. Furthermore, when peripheral blood mononuclear cells were employed as effector cells, the average antibody-dependent cellular toxicity (ADCC) activity exceeded 60%. In xenograft mouse models, this antibody treatment regimen has been shown to reduce the human CD45 + cell burden in the bone marrow and to extend the median survival of tumor-bearing mice from 17 to 28 days [45]. Although CLL-1 monoclonal antibodies have demonstrated ADCC/CDC activity in preclinical studies, their safety and efficacy have yet to be substantiated in human experimentation. In comparison with anti-CD33 antibodies, the antibody in question, despite being internalised by half within a period of half an hour and preferentially delivered to lysosomes, did not demonstrate a reduction in its effector function. Conversely, the accelerated antigen clearance diminished membrane epitope evasion, thereby establishing a theoretical framework for the subsequent clinical progression of antibody-drug conjugates (ADCs).
Antibody-drug conjugate (ADC)
ADC drug
CLL-1 is almost exclusively confined to the surface of leukemia stem cells and is negative on hematopoietic stem cells, providing an ideal target for ADCs, which can significantly reduce off-target toxicity [50]. Furthermore, the rapid internalisation of the latter provides a natural foundation for the delivery of highly active cytotoxic drugs. It is evident that research on monoclonal antibodies targeting CLL-1 is currently accelerating towards the development of ADCs. This has become a significant strategy for the therapeutic development of this target. To date, two CLL-1 ADCs targeting AML have been systematically evaluated:
The first compound is DCLL9718S, represents a first-generation ADC approach that encountered critical clinical limitations. It is a THIOMAB™ antibody-drug conjugate (TDC) consisting of a humanized monoclonal IgG1 anti-CLL-1 antibody (MCLL0517A) linked to two pyrrolobenzodiazepine (PBD) dimer drugs via a cleavable disulfide linker [51]. The PBD dimers function as DNA alkylating and cross-linking agents, forming covalent bonds with DNA in the minor groove upon release [11]. A Phase I dose-escalation trial in 18 adult patients with relapsed/refractory AML was prematurely terminated due to a lack of efficacy and significant toxicity [33]. The results demonstrated an objective response rate of zero, and as many as 67% of patients experienced treatment-related adverse events of grade 3 or higher. Notably, dose-limiting toxicity manifested as hepatic injury, a class effect observed with PBD-based payloads. The failure of DCLL9718S exemplifies the limitations of first-generation ADCs: suboptimal linker stability, heterogeneous drug-to-antibody ratio (DAR), and the intrinsic immunogenicity and rapid clearance associated with early conjugation technologies [52].
In contrast, CLT030 represents a next-generation ADC with optimized molecular architecture. Its composition includes a humanised IgG1 framework conjugated to the auristatin class microtubule inhibitor DUBA (a dolastatin-10 derivative) via a cleavable valine-citrulline linker with a PEG-8 spacer. This design incorporates two engineered cysteine residues for site-specific conjugation, achieving a homogeneous DAR of 1.7–1.8 with > 95% monomer purity. The valine-citrulline linker demonstrates superior plasma stability compared to the disulfide linker in DCLL9718S, as evidenced by maintained total antibody and ADC levels in human plasma for up to 5 days. Preliminary clinical studies indicate significant anti-leukemic properties: in vitro, CLT030 demonstrated picomolar IC₅₀ (0.5–27 ng/mL) against various AML cell lines (MOLM-13, OCI-AML2, HL-60), with killing efficiency positively correlated with target antigen abundance (R² = 0.87). At the primary cell level, treatment of 14 AML bone marrow samples resulted in > 80% reduction in leukemia colony-forming ability with limited impact on normal hematopoietic stem cells. In a primary xenograft model, a single intravenous dose of 0.3 mg/kg reduced peripheral blood human CD45 + cells by approximately 22-fold within 4 weeks, with bone marrow leukemia stem cells reduced below detection limits [46]. No significant weight loss or hematological toxicity was observed.
While CLT030 has demonstrated promising preclinical efficacy in AML models, its clinical development status remains unclear; no Phase I trial results have been published to date. This stands in contrast to DCLL9718S, which completed Phase I evaluation albeit with disappointing results. The lack of clinical progression for CLT030 may reflect the challenges faced by biotechnology companies in advancing ADC therapeutics, or strategic reprioritization following the demonstration of clinical proof-of-concept by other CLL-1-targeted modalities such as MCLA-117.
Optimization of ADC drugs
As a crucial strategy for achieving targeted therapy against CLL-1, the development of ADCs still faces multiple challenges. To reverse the termination of DCLL9718S due to high toxicity, subsequent optimization strategies can achieve breakthroughs in three core technologies: Drug to Antibody Ratio (DAR), linker chemistry, and payload selection.
Antibody Affinity and Specificity: First-generation ADCs like DCLL9718S employed conventional humanized antibodies without fine-tuned affinity modulation. Emerging evidence suggests that ultra-high-affinity antibodies may promote rapid internalization and lysosomal degradation, thereby narrowing the therapeutic window. Next-generation technologies employ site-specific conjugation techniques—including engineered cystine disulfide bonds, non-natural amino acid click chemistry, enzyme-catalyzed transamidation, and disulfide bond reconfiguration—to generate homogeneous ADCs with defined drug-to-antibody ratios (typically DAR 2) [53–55]. These approaches enhance ADC stability, reduce toxicity to normal myeloid cells (particularly CLL-1-expressing neutrophils), and enable higher dose administration.
Linker Stability and Hydrophilicity: The disulfide-linked linker in DCLL9718S exhibited insufficient plasma stability, leading to premature payload release and off-target toxicity [56, 57]. In contrast, CLT-030 employs a valine-cystine protease-cleavable linker and enhances hydrophilicity by introducing a PEG-8 spacer, thereby improving solubility and pharmacokinetic properties [46]. Third-generation ADCs further explore sulfonate self-destruct linkers and β-glucuronidase-cleavable linkers to enhance tumor selectivity [58].
Payload potency versus bystander effects: While the PBD dimer in DCLL9718S demonstrated potent efficacy, it exhibited significant hepatotoxicity—a limitation absent in the auristatin-based DUBA payload of CLT030 [46]. However, AML exhibits substantial antigenic heterogeneity, with refractory leukemic stem cells often displaying low or absent CLL-1 expression [59]. While CLL-1-targeted ADCs effectively eliminate CLL-1 positive cells, CLL-1 negative subpopulations may survive and drive relapse. To address this, next-generation ADCs employ membrane-permeable payloads (e.g., topoisomerase I inhibitor SN-38) to eliminate neighboring low-expressing or negative tumor cells via bystander killing effects [60]. This strategy directly counters antigenic heterogeneity challenges posed by CLL-1 expression variations across FAB subtypes, particularly in low-CLL-1-expressing M0 and M3 subtypes.
This technological evolution—from random conjugation using unstable linkers to site-specific, hydrophilic, stable linker-payload systems—reflects the overall advancement of ADC technology. First-generation ADCs exhibited issues such as high immunogenicity, unstable linkers, and inconsistent DAR values. While second-generation ADCs improved payload potency and linker stability, they retained random conjugation characteristics. Third/fourth-generation ADCs like CLT-030 integrate site-specific conjugation, optimized DAR, and hydrophilic linkers, representing the current frontier in CLL-1 targeted therapy. Future directions include developing dual-payload ADCs combining different mechanisms of action, such as microtubule inhibitors and DNA-damaging agents, to overcome resistance and prevent relapse caused by antigen escape [61].
Bispecific antibodies (BsAb)
Classification and mechanisms of bispecific antibodies
Bispecific antibodies (BsAbs) are a class of engineered antibodies that possess the ability to bind to two distinct antigens or epitopes in a simultaneous manner. The design of BsAbs overcomes the limitation of traditional monoclonal antibodies (mAbs) that target only a single antigen, achieving more complex therapeutic mechanisms through synergistic binding effects [62, 63], BsAbs have shown great potential in the treatment of hematological malignancies. Based on their mechanisms of action, BsAbs can be categorized into three major types: cell-bridging, dual-target inhibition, and immune modulation [64]. Cell-bridging BsAbs principally utilise one binding arm to recognise antigens on tumor cell surfaces, such as CLL-1, CD33, and CD123 on AML cells, whilst the other arm recognises activating receptors on the surface of immune effector cells. The formation of an immunological synapse between tumor and immune cells has been demonstrated to facilitate the specific elimination of tumor cells by immune cells [65–67], A notable example of this phenomenon is the utilisation of Blinatumomab, a drug that is extensively employed in the treatment of acute lymphoblastic leukaemia (ALL) [68]. Furthermore, these antibodies have been observed to induce the secretion of pro-inflammatory cytokines, such as interferon-γ and tumor necrosis factor-α. These, in turn, have been shown to inhibit the proliferation of leukaemia cells, activate macrophages and dendritic cells in a coordinated manner, and reshape the bone marrow microenvironment [69]. Furthermore, interferon-γ has been shown to upregulate MHC-I expression on tumor target cells, thereby indirectly enhancing adaptive immune recognition and further exerting anti-tumor effects [70], The concept of dual-target inhibition BsAbs refers to antibodies that simultaneously bind to two different signalling molecules or receptors on the same cell, thereby blocking two independent signalling pathways at the same time. This is believed to prevent compensatory activation that may occur subsequent to single-target inhibition. By retaining Fc-mediated ADCC/ADCP, these antibodies enhance the cytotoxic effects [71]. Examples of such dual receptor inhibition include that of EGFR × IGF-1R, PDGFRα × PDGFRβ, and receptor-ligand dual inhibition of VEGF × DLL4 [72, 73].The focus of immune modulation BsAbs is twofold, targeting both tumor-associated antigens (TAAs) and immune checkpoint molecules, including PD-1/PD-L1 and CTLA-4 [74]. Despite their present lesser application in the context of hematological malignancies, these agents have demonstrated significant efficacy in the treatment of solid tumors. The mechanism of action of these agents involves the concentration of the drug in the tumor microenvironment(TME) through the TAA arm, thereby effectively reducing systemic toxicity caused by peripheral immune activation. Furthermore, by obstructing inhibitory signals via the checkpoint arm, they directly alleviate T-cell functional suppression. This dual action engenders a high-concentration immune-stimulating environment, locally activating CD8 + T cells and depleting immunosuppressive regulatory T cells, thereby reshaping the tumor immune microenvironment and enhancing anti-tumor immune killing [75]. Examples of this include PD-L1 × CD3, PD-L1 × CTLA-4 bispecific antibodies [76–78].
Bispecific antibodies targeting CLL-1
The development strategy for CLL-1-targeted BsAbs is currently focused on cell-bridging BsAbs with a CLL-1/CD3 dual-target design. MCLA-117, a pioneering bispecific antibody, has been developed to target CD3 on T cells and CLL-1 on leukemia cells. This results in the redirection and activation of T-cell subsets in a manner that is independent of MHC or costimulatory signals. Van Loo et al. demonstrated that 10 ng/mL of MCLA-117 can induce > 90% lysis of primary AML blasts, with an EC50 range of 0.5–2.5 ng/mL, significantly lower than that of the CD33×CD3 control antibody (EC50 20–50 ng/mL) [79]. In mouse models, a single intravenous injection of 0.5 mg/kg MCLA-117 reduced the level of human CD45 + cells in the bone marrow to the detection limit (< 0.1%) within 7 days, while the control group remained between 60 and 80%; no grade 2 or higher cytokine release syndrome(CRS) or significant weight loss was observed. Subsequent toxicology studies conducted on cynomolgus monkeys demonstrated that, even at the maximum dose of 10 mg/kg, there was no persistent T-cell depletion or substantial organ pathology, thereby establishing a safety foundation for the drug to enter first-in-human clinical trials [34]. Furthermore, ABL602 is a cell-bridging BsAb with an asymmetric 2 + 1 architecture, comprising two CLL-1 Fabs and one CD3ε Fab. This results in a reduction of CD3 affinity to approximately 60nM. The weak CD3 binding forces the antibody to first form a high-concentration platform on tumor cells via bivalent CLL-1, followed by local accumulation of multiple weak signals to activate T cells, thereby avoiding excessive peripheral activation. In vitro experiments demonstrated that ABL602 has an EC50 of 0.05–0.3 ng/mL for primary AML, which is comparable to that of a potent control antibody. However, it exhibited a 60–80% reduction in the peak levels of IFN-γ, IL-6, and TNF-α (P < 0.01). In an NSG mouse model with primary AML-PBMCs, a dose of 0.02 mg/kg (administered twice weekly) reduced human CD45 + cells in the bone marrow by more than two logs. The ABL602 group exhibited stable body weight, and no grade > 2 toxicity was observed. In cynomolgus monkeys, a dose of 10 mg/kg resulted in only transient T-cell depletion (≤ 30%, recovering within 48 h) without persistent organ damage, supporting its advancement to clinical trials [80], These findings demonstrate the therapeutic potential of CLL-1 -targeted BsAbs in the field of AML. The current research results for CLL-1 targeted BsAbs are summarised in Table 2.
Limitations and optimization strategies for CLL−1 -targeted BsAbs
Despite the considerable promise of bispecific antibodies (BsAbs) targeting CLL-1 for the treatment of AML, there are several significant challenges and limitations to be addressed. The most critical challenge is that of target-related toxicity. Despite the absence of CLL-1 expression in hematopoietic stem cells, its presence has been documented on normal myeloid progenitor cells, monocytes, and granulocytes. Consequently, treatment invariably results in myeloid suppression [12]. Tumor heterogeneity and antigen escape have been identified as significant factors contributing to the failure of BsAb therapy [81]. Additionally, the inhibitory cytokines secreted by regulatory T cells and myeloid-derived suppressor cells have the capacity to weaken the function of effector cells, thereby limiting the efficacy of BsAbs [67]. Despite the fact that CLL-1 targeted BsAbs (for example, ABL602) have reduced CD3 affinity in order to mitigate CRS, CRS remains a significant risk that must be continuously balanced in T-cell-redirecting therapies.
In order to address these challenges, the field is actively exploring various strategies to enhance efficacy and overcome these bottlenecks. In order to address treatment-related myeloid suppression, there is a necessity to establish standardised supportive care protocols. Such protocols should include the prophylactic use of granulocyte colony-stimulating factors and other supportive therapies for active management. In order to enhance efficacy, a combination of BsAbs with hypomethylating agents has been demonstrated to alter the tumor epigenetic state, increase target antigen density, and improve the microenvironment [82, 83]. Furthermore, the construction of dual-target molecules that target both CLL-1 and other antigens can effectively prevent relapse due to the loss of a single antigen. In molecular design, the optimisation of pharmacokinetic properties and activation strength through Fc segment genetic engineering is intended to achieve an enhanced therapeutic window [84], Consequently, a comprehensive investigation of the aforementioned strategies will furnish a substantial theoretical foundation for overcoming the application limitations of BsAbs.
Table 2. Bispecific Antibody (BsAb) Studies Targeting CLL-1DrugTargetsFormatIn Vitro Efficacy(EC50)Key Translational EfficacyStudy StageFirst-in-Human TrialCLL1-TriKE [85]CD16a×IL-15 × CLL-1TriKE(NK engager)Not disclosedInduces NK-cell killing of primary AML/LSCs at 0.5–5 ng/mL; significantly reduces leukemic burden in NSG mouse BM with ≤Grade 2 toxicity.PreclinicalNot yet initiatedMCLA-117 [34]CLL-1 ×CD32:1 IgG4 BsAb68 ± 37 ng/mL (HL60)> 99% leukemia clearance; targets primary AML cells and LSCsPhase INCT03038230ABL602 [80]CLL-1 × CD3T-cell engager (2:1)0.05–0.3 ng/mL> 2-log leukemia burden reduction at low dose (0.02 mg/kg)PreclinicalNot yet initiatedCLEC12A-ENG / CLEC12A-ENG.CD123IL7Rα (DART) [86]CLL-1×CD3 + CD123×IL-7RαDART-ENG (dual)0.8 ng/mL> 95% leukemia clearance; dual-targeting strategy reduces relapse riskPreclinicalNot yet initiatedQL325CLEC12A×CD32 + 1 valent BsAbNot disclosedDose-dependent AML suppression in vivoPreclinicalNot yet initiatedCD3 × CLL-1 [87]CD3×CLL-12:1 IgG4-like BsAbNot disclosedComplete tumor elimination with single dose (1 mg/kg)PreclinicalNot yet initiatedBsAb bispecific antibody, AML Acute Myeloid Leukemia, NK Natural Killer (cell), LSC Leukemia Stem Cell, BM Bone Marrow, DART Dual Affinity Re-targeting Technology, ENG Engineered
Trispecific antibodies (TsAbs)
Trispecific antibodies (TsAbs) are engineered to bind to three different antigens, typically including two tumor-associated antigens (TAAs) and one immune cell activation receptor. Examples of such receptors include CD19/CD3/CD20, CD33/CD3/CD123, CD123/NKp46/CD16a, and CD16/IL-15/CD33 (TriKE) [88]. Through this multivalent binding, TsAbs direct immune cells to kill AML cells. In comparison with BsAbs, TsAbs have been shown to reduce escape due to a single target. The incorporation of a third domain, such as IL-15, into TsAbs has been demonstrated to prolong the half-life, facilitate costimulatory signals, and enhance stability [69, 70, 89], thereby achieving dual stimulation signals. Furthermore, TsAbs, through a “weak CD3 and strong tumor bivalent” design, ensure that immune activation occurs only in high antigen density microregions, thereby reducing the risk of CRS.
Arvindam et al. constructed CLL-1-TriKE [85], which uses a CD16a-IL-15-CLEC12A trivalent structure to simultaneously anchor natural killer (NK) cells and AML cells. The IL-15 domain has been demonstrated to amplify the signal autonomously, thereby enabling strong lysis to be triggered at low concentrations. This molecule has been shown to clear over 90% of primary AML and LSC in vitro at concentrations ranging from 0.5 to 5 ng/mL, with significant upregulation of NK degranulation and IFN-γ secretion. In vivo, a dose of 0.1 mg/kg reduced the human CD45 + burden in the bone marrow of NSG mice by more than two logs, with LSC nearly at the detection limit, and no grade > 2 toxicity throughout the process. This finding serves to substantiate the hypothesis that the substance is characterised by elevated efficiency and minimal toxicity, thereby establishing a preclinical foundation for its subsequent clinical translation. Despite the fact that TsAbs are still in the early exploration stage, their innovative design concept has opened up a new treatment pathway for patients with relapsed/refractory (R/R) AML, especially for those who have failed BsAb or CAR-T therapies. By meticulously selecting tumor target antigens, optimising the Fc segment structure, and precisely regulating the intensity of immune activation, TsAbs are anticipated to attain an ideal equilibrium between efficacy and safety in the future, thereby providing a more efficient and tolerable immunotherapy option for clinical utilisation.
CAR-T cells anti-CLL-1
Chimeric Antigen Receptor T-cell (CAR-T) therapy is a state-of-the-art immunotherapy technology that involves the genetic engineering of T cells to express specific antigen receptors, thereby enabling them to precisely identify and destroy tumor cells [90, 91]. CAR-T therapy has achieved considerable success in the treatment of B-cell malignancies and plasma cell tumors [92, 93], however, its application in AML has been limited by the paucity of suitable targets. CLL-1 is expressed at high levels on over 90% of AML cells, but is either not expressed or expressed at very low levels on normal hematopoietic stem cells. This makes it an ideal antigen target following CD33 and CD123 [94, 95].
Jinghua Wang et al. [96]constructed a second-generation CLL-1 CAR-T that can efficiently and selectively lyse AML blasts and leukemia stem cells in vitro, with no toxicity to normal CD34 + CD38- haematopoietic stem cells. A single dose of 1 × 10⁶ CAR-T cells reduced the leukemia burden in the bone marrow of NSG mice by more than two logs, with no deaths related to CRS or weight loss. This preclinical data established a robust foundation of high efficiency and safety for the clinical translation of CLL-1-targeted CAR-T therapy for relapsed/refractory (R/R) AML. Jin X et al. [33]were the first to evaluate the feasibility of CLL-1-targeted CAR-T cell therapy for R/R AML in humans. This Phase I clinical trial (NCT04388944) enrolled 10 adult patients who had failed multiple lines of therapy. The results demonstrated that following fludarabine-cyclophosphamide lymphodepletion, the infusion of 1–2 × 10⁶ cells/kg of CAR-T cells resulted in an objective response rate of 70%, with three patients attaining minimal residual disease (MRD)-negative complete remission. Furthermore, CLL-1⁺ cells in the bone marrow were undetectable within 28 days, and CAR-T persistence was > 6 months. With regard to safety, all patients experienced CRS, with six cases being high-grade, but no CRES was observed. The principal dose-limiting toxicity was identified as prolonged neutropenia. This study was the first to confirm in humans that CLL-1 CAR-T therapy can induce deep and durable clinical remission, with controllable safety but a need for further optimization of myelosuppression. This study thus lays an important foundation for subsequent clinical development. It is particularly noteworthy that for patients with AML accompanied by extramedullary diseases (EMDs), CLL-1 CAR-T therapy also demonstrated satisfactory efficacy. A clinical investigation (ChiCTR2000041054) [97], revealed that, in a cohort of 20 AML patients with EMDs, the complete remission rate of CLL-1 CAR-T therapy was 65.00%, and that these patients subsequently achieved long-term survival through the process of haematopoietic stem cell transplantation. Current CAR-T treatments targeting CLL-1 are steadily advancing, as evidenced in Table 3, and may provide a new therapeutic strategy to improve the clinical outcomes of patients with R/R AML.
Notwithstanding the encouraging outlook for CLL−1 CAR-T therapy in the treatment of AML, there are numerous challenges to be surmounted before its clinical translation can be realised. In a phase I trial of a chimeric antigen receptor (CAR) T-cell therapy for paediatric relapsed/refractory (R/R) acute lymphoblastic leukaemia (ALL), three patients achieved short-term complete remission. However, the third patient relapsed molecularly on day 30. The results of the flow cytometry analysis indicated that the newly identified leukemia clusters predominantly exhibited a negative expression of the CLL−1 antigen. This finding indicates that while CLL−1 CAR-T therapy can indeed induce remission, its efficacy is transient, and antigen loss emerges as a pivotal mechanism through which leukemia stem cells (LSCs) evade therapy [59]. In order to overcome the phenomenon of antigen escape, the use of multi-target combined CAR-T is indicated. For instance, CD33-CLL−1 CAR-T and CD123-CLL−1 CAR-T utilise a tandem CAR configuration to target two distinct antigens. A study was conducted that targeted CD33 and CLL−1 [35], The study revealed that CD33-CLL−1 CAR-T exhibited a high level of efficacy in eradicating low or negative AML cells (e.g., MOLM−13) in vitro. In a mouse model, CD33-CLL−1 CAR-T significantly reduced tumor load and extended mouse survival. In human experimentation, Liu F’s team employed a P2A self-cleaving peptide to construct a dual-target cCAR. In this study, the patient was a child suffering from relapsed R/R AML (73% PB, 81% BM blasts). The conditioning regimen consisted of fludarabine and cyclophosphamide (Flu/Cy), followed by a two-day infusion of 1 × 10⁶ cCAR-T/kg. On the twelfth day, there was a temporary rise in bone marrow blasts to 98%, but there was significant expansion of CAR-T cells. By the nineteenth day, the patient had achieved MRD-negative CR with 36% PB and 60% BM CAR-T. Thereafter, the child underwent non-myeloablative allo-HSCT, which resulted in a significant reduction in toxicity. This case demonstrates the potential of CD33-CLL−1 cCAR to effectively overcome multidrug resistance, achieving deep remission and establishing a safe transplant window. In addition to dual antigen targets, the combination of CLL−1 CAR-T with epigenetic regulators such as decitabine and panobinostat has been shown to reverse CLL−1 expression in certain AML cells, thereby restoring CAR-T recognition [98].
Toxicity associated with CLL−1 CAR-T therapy is also one of the core challenges in clinical translation. In addition to common CRS and ICANS, myelosuppression and infection are prevalent adverse reactions [99, 100]. Consequently, the potential impact of CLL−1 CAR-T on CLL−1-positive normal cells must be closely monitored and meticulously managed, despite the relatively limited expression of CLL−1 in normal tissues.
Table 3. Clinical Trials of Anti-CLL−1 CAR-T Therapy in Acute Myeloid LeukemiaNCT/ChiCTR NumberSimplified Therapeutic Approach(Modified)Target(Modified)Population(Modified)Phase & StatusKey Translational FeaturesACTIVE/RECRUITING STUDIESNCT07201727CD123 + CLL-1 Sequential CAR-T → Allo-HSCT bridgeCD123 + CLL-1R/R AMLEarly Phase 1Not yet recruitingSequential dual-targeting + stem cell transplant bridge; curative strategyNCT07198867A-CAR028 (CLL-1 CAR-T)CLL-1R/R AMLPhase 1RecruitingNovel single-target constructNCT07036250U32 CAR-TCLL-1AMLPhase 1/2Recruiting-NCT06917105CD33/CD123/CLL-1 Triple CAR-TCD33 + CD123+CLL-1Myeloid MalignanciesPhase 1/2Not yet recruitingTriple-targeting to address clonal heterogeneityNCT06880354CLL-1/CD38 Dual CAR-TCLL-1 + CD38AMLPhase 1RecruitingDual-targeting; CD38 adds tumor coverageNCT06680752ARD103 CAR-T + Flu/CyCLL-1R/R AML, R/R MDSPhase 1/2RecruitingExtends to MDS; combination with chemotherapyNCT06347458BG1805CLL-1R/R AML (Pediatric)Phase 1Not yet recruitingPediatric focusChiCTR2500100811CLL-1 CAR-TCLL-1AMLRetrospectiveRecruitingRetrospective analysis of existing dataNCT06118788BG1805CLL-1R/R AMLPhase 1/2RecruitingAdult population (companion to pediatric NCT06347458)NCT06027853CLL-1 CAR-NKCLL-1Adult AMLPhase 1RecruitingNK cell platform (not T-cell); off-the-shelf potential; reduced CRS riskNCT06017258CD371-YSNVz/I-18 CAR-TCD371 (CLL-1)AMLPhase 1RecruitingNovel construct with IL-18 engineeringNCT05995041Universal CAR-T (Multi-target)CLL-1/CD33/CD38/CD123AMLPhase 1RecruitingUniversal/allogeneic platform; quadruple targetingNCT04219163CLL-1 CAR-TCLL-1AMLPhase 1Active, not recruiting-TERMINATED/WITHDRAWN STUDIESNCT06128044CB-012 (CRISPR-edited Allogeneic CAR-T)CLL-1R/R AMLPhase 1TerminatedGene-edited universal cells (discontinued; important negative data)NCT06110208CLL-1/CD38 Dual CAR-TCLL-1 + CD38AMLEarly Phase 1TerminatedDual-targeting (terminated)NCT05943314CLL-1/CD33 CAR-TCLL-1 + CD33AMLPhase 1WithdrawnDual-targeting (withdrawn)NCT05654779LCAR-AMDR (CLL-1/CD33)CLL-1 + CD33R/R AMLPhase 1TerminatedDual-targeting with AMDR modification (discontinued)NCT05248685Dual CD33/CLL-1 CAR-TCD33 + CLL-1R/R AMLPhase 1WithdrawnDual-targeting (withdrawn)UNKNOWN STATUS STUDIESChiCTR1900028303CLL-1 CAR-TCLL-1AMLPhase 1Unknown-ChiCTR2000036350Multi-target CAR-T (incl. CLL-1)CD19/20/22/33/123/CLL-1/138/BCMA/CD30AMLPhase 1UnknownPan-hematologic targeting strategyNCT05467254CLL-1 + CD33 CAR-TCLL-1 + CD33R/R AMLPhase 1UnknownDual-targetingNCT05467202CLL-1 CAR-TCLL-1R/R AMLPhase 1Unknown-NCT05252572CLL-1 CAR-TCLL-1AMLEarly Phase 1Unknown-NCT05016063Dual CD33-CLL-1 CAR-T + Flu/CyCD33 + CLL-1R/R AMLEarly Phase 1UnknownDual-targeting with lymphodepletionNCT04923919Anti-CLL-1 CAR-TCLL-1R/R AMLEarly Phase 1Unknown-NCT04884984Anti-CLL-1 CAR-TCLL-1R/R AMLPhase 1/2Unknown-NCT04010877Multi-CAR-T (CLL-1/CD33/CD123)CLL-1 + CD33+CD123AMLPhase 1/2UnknownTriple-targetingNCT03795779CLL-1-CD33 cCARCLL-1 + CD33R/R AML, MDS, MPN, CMLEarly Phase 1UnknownDual-targeting; broad myeloid indicationADVANCED PHASE STUDIESNCT03631576CD123/CLL-1 CAR-T (STPHI_0001)CD123 + CLL-1R/R AMLPhase 2/3UnknownMost advanced; dual-targeting in late-stage developmentNCT03222674Multi-CAR-T (Muc1/CLL-1/CD123 etc.)MultipleAMLPhase 1/2UnknownMulti-antigen approachAML Acute Myeloid Leukemia, MDS Myelodysplastic Syndrome, R/R Relapse/Refractory, MPN Myeloproliferative Neoplasm, CML Chronic Myeloid Leukemia
CLL-1 CAR-NK
CAR-NK cells are a type of natural killer (NK) cell that has been genetically modified. The core feature of these cells is the presence of a CAR on their surface, which has the capacity to bind specifically to certain antigens on the AML cell membrane with a high degree of specificity [101, 102]. The fundamental structure encompasses three primary domains: the extracellular antigen recognition domain with a single-chain variable fragment (scFv) targeting the antigen, the transmembrane domain, and the intracellular signaling domain [103, 104]. The CLL-1 CAR-NK cells have been shown to specifically target CLL-1-positive AML cells. Upon contact, the scFv of the CLL-1 antigen on the CAR induces the formation of an immunological synapse. The intracellular signalling modules, such as CD3ζ and 4-1BB/CD28, in the CAR-NK cells rapidly cluster [105], thereby triggering a robust activation cascade. This cascade prompts the NK cells to swiftly transition from a quiescent state to an effector state, thereby acquiring high-efficiency killing capabilities and exerting anti-tumor effects.
Preclinical studies of CLL-1 CAR-NK have achieved key breakthroughs. Pioneering work by Gurney et al. first established CLL-1 CAR in primary NK cells using a non-viral transposon system, while simultaneously knocking out the CISH gene regulating the IL-15 signaling pathway via CRISPR/Cas9 technology [106]. This modification not only significantly enhanced the in vitro expansion capacity and cytotoxic efficacy of CAR-NK cells against AML cell lines, patient primary cells, and LSCs, but also extended median survival from 29 days to over 90 days in primary AML patient-derived xenograft (PDX) mouse models. Some mice achieved potential cure, providing robust evidence for clinical application.
Recently, further engineering strategies have focused on overcoming the bottleneck of insufficient in vivo persistence in traditional CAR-NK cells. These “armored” designs primarily include: (1) co-expressing IL-15 (membrane-bound or secreted) to autonomously provide survival signals, maintaining cell viability and a memory-like phenotype; (2) Dual CAR designs (e.g., simultaneously targeting CLL-1 and CD33) to counteract antigenic heterogeneity in AML and reduce the risk of antigen escape. The 2024 American Society of Hematology (ASH) Annual Meeting reported that CLL-1 CAR-NK cells fortified with “structural optimization + membrane-bound IL-2” demonstrated enhanced in vivo leukemia clearance and prolonged survival benefits in preclinical models. The corresponding first-in-human trial is now in preparation. Additionally, induced pluripotent stem cell (iPSC)-derived CAR-NK platforms (e.g., FT596) offer the potential for standardized, scalable “off-the-shelf” products due to their unlimited expandability and uniform quality, representing a significant frontier in CLL-1 targeted therapy [107].
Compared to CAR-T, CLL-1 CAR-NK possesses multiple potential advantages. CAR-NK cells do not rely on HLA recognition, and allogeneic infusion rarely induces graft-versus-host disease(GVHD) [108]. Their activated cytokine profile primarily consists of IFN-γ and GM-CSF, with minimal production of IL-6 and other cytokines associated with severe CRS. Consequently, the risk of ≥Grade 3 CRS and immune effector cell-associated neurotoxicity syndrome (ICANS) is significantly reduced [109]. Furthermore, while retaining CAR-specific killing, CAR-NK cells preserve innate NK cell functions (e.g., recognition via receptors like NKG2D and DNAM-1, and ADCC), enabling clearance of tumor cells with downregulated target antigens and reducing relapse risk. Benefiting from their inherent allogeneic safety profile, CAR-NK products can be manufactured in advance and cryopreserved, enabling immediate patient access. This addresses core challenges of autologous cell therapies, including lengthy cycles, high costs, and suboptimal cell quality in some patients.
Currently, multiple preclinical and early-stage clinical studies are actively advancing the use of CLL-1 CAR-NK therapy for R/R AML (Table 4). The primary objectives are to validate safety and achieve long-lasting efficacy in vivo, thereby providing AML patients with a novel, highly effective, safe, and readily accessible immunotherapy option.
Table 4. Clinical Study of anti CLL-1 CAR-NK Cell Therapy for AMLNCT NumberTherapeutic ApproachPopulationPhase & StatusKey Translational FeaturesNCT06367673iPSC-derived CAR-NK cells(CLL−1 or CD33 targeted)Adult AMLPhase 1RecruitingiPSC platform: scalable manufacturing, true off-the-shelf potential; dual-targeting capability (CLL−1/CD33 switch)NCT06307054CLL−1 CAR-NK cellsR/R AMLPhase 1RecruitingCAR-NK platform: inherent safety profile (reduced CRS/ICANS risk vs. CAR-T); no requirement for patient-specific manufacturingNCT06027853CLL−1 CAR-NK(cell injection)Adult AMLPhase 1RecruitingDirect CAR-NK administration; standard NK cell engineering approachNCT05215015CD33/CLL−1 Dual CAR-NKAMLEarly Phase 1UnknownDual-targeting strategy: simultaneous CD33 + CLL−1 targeting to prevent antigen escape and relapseR/R AML Relapsed/Refractory Acute Myeloid Leukemia, CRS Cytokine Release Syndrome, ICANS Immune Effector Cell-Associated Neurotoxicity Syndrome, CAR-NK Chimeric Antigen Receptor-Natural Killer
Neutropenia and management
Targeted CLL-1 therapy induces granulocytopenia due to the “targeted/tumor-depletion” effect of targeted treatment on normal myeloid progenitor cells and mature neutrophils, clinically manifesting as widespread and severe pancytopenia. Particularly in CAR-T therapy, the persistent in vivo survival and function of CAR-T cells continuously eliminate newly generated CLL-1-expressing myeloid cells. This leads to long-term, treatment-dependent neutropenia and may even trigger life-threatening infections. Neutropenia is a core risk factor for infection in cancer patients [110]. Following CAR-T therapy targeting CLL-1, nearly all patients experience severe pancytopenia, placing them at extremely high risk of infection [32, 33]. On one hand, CAR-T cells directly recognize and eliminate circulating and bone marrow-resident neutrophils and their progenitor cells expressing CLL-1. On the other hand, intense CRS accompanied by high levels of inflammatory cytokines (such as IL-6 and IFN-γ) may further suppress the bone marrow hematopoietic microenvironment, impairing recovery of all blood cell lines and prolonging the duration of cytopenia [111].
Management strategies for neutropenia should encompass a multidimensional approach covering “monitoring-prevention-intervention.” Supportive care forms the foundation for managing prolonged neutropenia, aiming to prevent and address complications. For high-risk patients with pre-existing neutropenia, prophylactic antimicrobial therapy should be considered. This includes prophylaxis against bacteria, fungi, and viruses. For patients developing granulopenia, consider granulocyte colony-stimulating factor (G-CSF) to promote neutrophil recovery. However, G-CSF use in the CAR-T therapy context requires caution, balancing its benefits in promoting myeloid recovery against the potential risk of exacerbating CRS.
Since residual CAR-T cells continuously eliminate newly emerging CLL-1-positive cells, G-CSF support alone is unlikely to achieve sustained hematopoietic recovery. Therefore, employing CLL-1 CAR-T therapy as a “bridge” strategy following deep clearance of leukemic cells to facilitate allogeneic hematopoietic stem cell transplantation (allo-HSCT) has emerged as the most effective clinical management pathway: transplanting donor hematopoietic systems that do not express this CAR resolves long-term granulocytopenia while leveraging graft-versus-leukemia effects to consolidate efficacy and prevent relapse [32, 33, 112, 113]. Studies have confirmed that bridging allo-HSCT after CLL-1 CAR-T-induced remission successfully achieves complete donor cell chimerism and hematopoietic reconstitution, thereby facilitating long-term survival for patients.
Immunosuppressive tumor microenvironment
Beyond antigen escape, systemic immune suppression mediated by the TME represents a core challenge limiting the efficacy of CLL-1 targeted therapies. The AML bone marrow microenvironment exhibits highly immunosuppressive characteristics, manifested by enrichment of immunosuppressive cell populations such as regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), along with high concentrations of immunosuppressive cytokines like IL-10 and TGF-β. The microenvironment exhibits a state of hypoxia, nutrient deprivation, and elevated metabolic waste (lactate, adenosine) [114]. It drives T-cell exhaustion by downregulating pro-inflammatory cytokine secretion (e.g., IFN-γ) and upregulates immune checkpoint molecules (PD-1, LAG-3, TIM-3), thereby suppressing antitumor activity of T cells and NK cells [115, 116].
Current therapeutic strategies targeting CLL-1 generally lack intrinsic mechanisms to counteract this immunosuppressive microenvironment. Although CLL-1-TriKE effectively induces NK cell activation and IFN-γ production, its mechanism is limited to direct targeting and killing, unable to neutralize inhibitory signals like TGF-β or IL-10 within the microenvironment. Similarly, CLL-1 CAR-T/NK cells are susceptible to metabolic stressors like hypoxia, nutrient deprivation, and lactate accumulation, leading to impaired cell function and insufficient persistence. This vulnerability to microenvironmental suppression underscores the necessity of combination therapy strategies for achieving durable remission. Currently, overcoming TME immunosuppression can be approached through multiple dimensions: (1) Constructing “armored” CAR structures enables effector cells to constitutively secrete pro-inflammatory cytokines like IL-12, IL-15, and IL-18. This suppresses Treg activity and enhances T/NK cell cytotoxicity, effectively countering IL-10/TGF-β-mediated immunosuppression. Additionally, CRISPR/Cas9 gene editing to knockout inhibitory receptors like TGF-β receptor II (TGF-βRII) or NKG2A confers direct resistance to TGF-β in CAR-T/NK cells, significantly enhancing their survival and function within suppressive microenvironments. (2) Combination with immune checkpoint inhibitors: The PD-1/PD-L1 pathway represents a critical immune evasion mechanism in AML, with PD-L1 expression on leukemic blasts strongly correlated with poor prognosis [117]. Combining CLL-1-targeted therapies with PD-1/PD-L1 inhibitors (e.g., nivolumab, pembrolizumab) synergistically enhances antitumor immune responses by reversing T-cell exhaustion and restoring cytotoxic function. Clinical studies demonstrate that combining hypomethylating agents (HMAs, e.g., azacitidine) with PD-1 blockade in R/R AML yields significantly improved response rates [118], suggesting synergistic potential when this combination strategy is used alongside CLL-1-targeted therapies. (3) Targeting metabolic reprogramming to reshape the TME: Modulating glucose metabolism, amino acid metabolism (e.g., arginine, tryptophan), and lipid metabolism within the microenvironment reduces the production of immunosuppressive metabolites like lactate and adenosine, thereby alleviating TME-mediated immune suppression [119–121]. For example, targeting lactate dehydrogenase-A (LDH-A) to lower lactate concentrations helps reverse the impairment of effector cell function caused by low pH environments [122], thereby enhancing the efficacy of anti-CLL-1 therapy.
In summary, the AML bone marrow microenvironment establishes profound immunosuppression through cellular, soluble factor, and metabolic mechanisms, severely limiting the durable efficacy of CLL-1 targeted therapies. Future clinical development should prioritize combination regimens integrating these multidimensional strategies. By enhancing effector cell adaptability, releasing checkpoint inhibition, and reshaping metabolism, comprehensive microenvironment normalization and long-term disease control can be achieved.
Conclusion
CLL-1 has emerged as a crucial target for precision therapy in AML due to its highly specific expression in LSC and near absence in normal hematopoietic stem cells. The transmembrane receptor structure of the subject is notable for its propensity for rapid internalisation, a property that renders it suitable not only for antibody-mediated cytotoxic reactions, but also for the efficient delivery of drugs or toxins. In recent years, a variety of strategies, including monoclonal antibodies, ADC, BsAbs and trispecific antibodies, and emerging CAR-T and CAR-NK cell therapies, have explored CLL-1 as a target. The findings from preclinical and early clinical trials have provided substantial evidence to support the hypothesis that the substance in question possesses both high specificity and anti-leukaemia activity. Nevertheless, ongoing development must address challenges such as immune escape, heterogeneous expression, an immunosuppressive bone marrow microenvironment, myelosuppression, and CRS/ICANs. It is recommended that future research endeavours concentrate on the synergistic design of other targets, including CD33 and CD123. The employment of dual or trispecific constructs, in conjunction with domain optimisation, has been demonstrated to enhance the sustained killing power of the system. The combination of targeted therapies directed at the CLL-1 antigen with IL-15, or other immune-modulating factors, has the potential to enhance tolerability. The integration of multiple platforms and the adoption of personalised strategies have been identified as key factors in the development of targeted treatments for patients with R/R AML. These treatments have the potential to offer safer, more precise, and sustainable options, representing a significant advancement in the field of cancer treatment.