Large-scale metagenomic surveillance study expands the known diversity of RNA viruses in mosquito populations from the Amazon Basin
Eddie Fuques, Aimee L. Massey, Faris Qureshi, João Vitor Campos-Silva, David J. Ferreira da Silva, Carlos A. Peres, Taal Levi, Rebecca L. Vega Thurber

TL;DR
This study used metagenomics to discover over 500 RNA viruses in mosquitoes from the Amazon Basin, revealing many new species and expanding our understanding of viral diversity in this region.
Contribution
The study presents one of the most comprehensive surveys of RNA viruses in Amazonian mosquitoes, identifying 18 putative novel viral species.
Findings
Over 500 viral sequences from 18 families were identified, including 21 complete or nearly complete genomes.
Eighteen putative novel viral species were discovered across diverse virus families.
Undocumented viral diversity was found in several families, including Iflaviridae and Togaviridae.
Abstract
The Amazon Basin is one of the most biologically diverse regions on Earth, yet its viral diversity remains poorly characterized. Mosquitoes are important vectors and reservoirs of RNA viruses, but little is known about the composition and structure of their viromes in remote areas of the Amazon. In this study, we performed a large-scale metagenomics survey of RNA viruses associated with mosquito populations collected from the Jurua River region in the Western Amazon Basin of Brazil. We analyzed 211 pooled samples of adult female mosquitoes collected across thirty-seven sites, representing one of the most comprehensive mosquito virome studies conducted in this region to date. Utilizing high-throughput sequencing and de novo assembly, we identified over 500 viral sequences from 18 families, including 21 complete or nearly complete genomes. Our analysis revealed 18 putative novel viral…
Click any figure to enlarge with its caption.
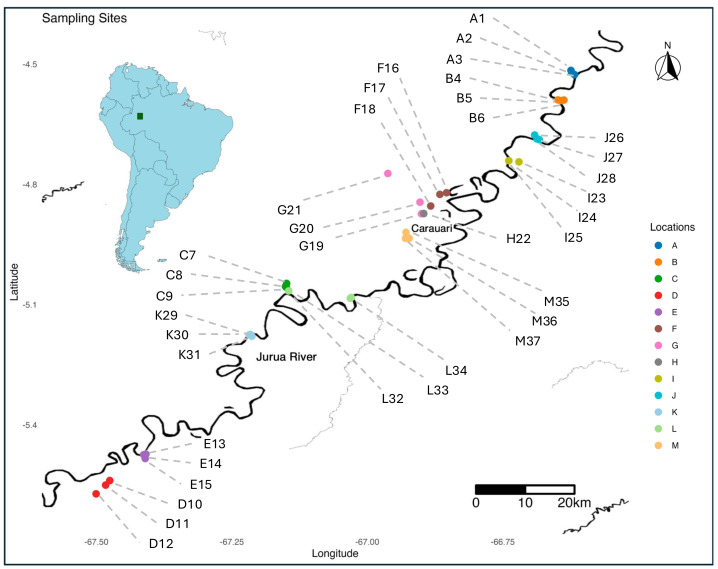 Figure 1
Figure 1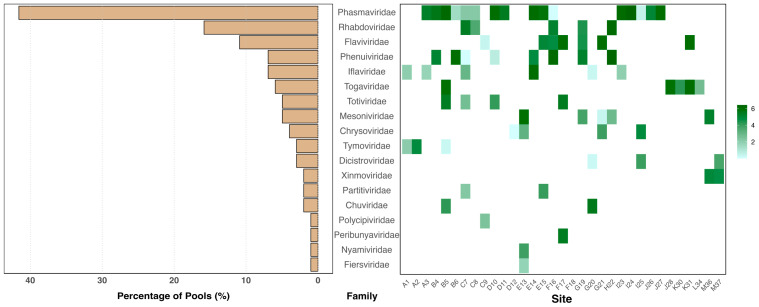 Figure 2
Figure 2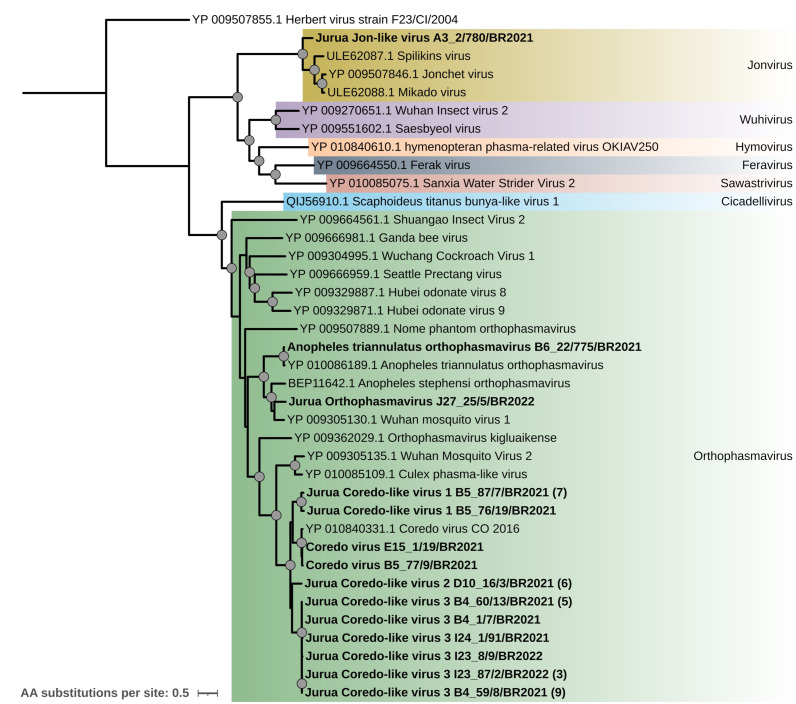 Figure 3
Figure 3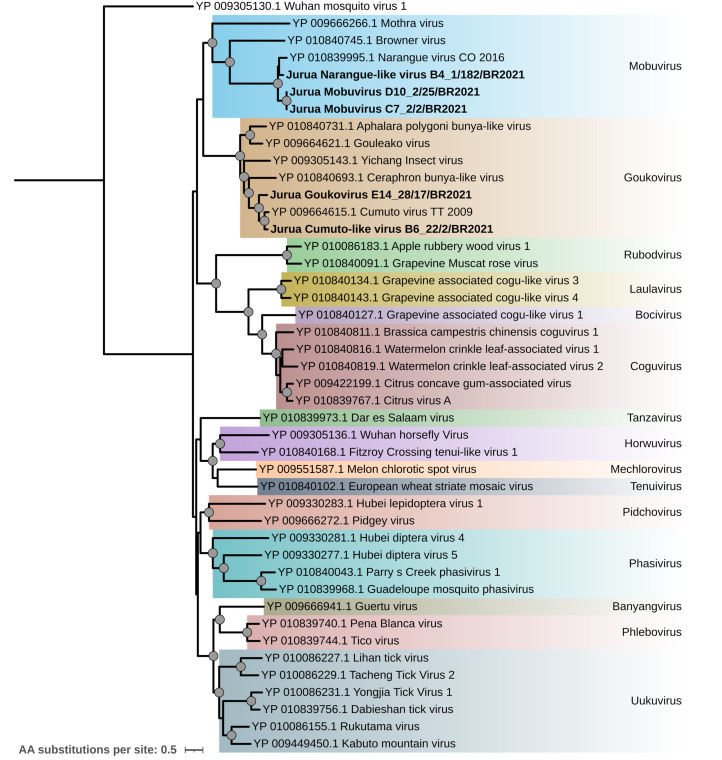 Figure 4
Figure 4 Figure 5
Figure 5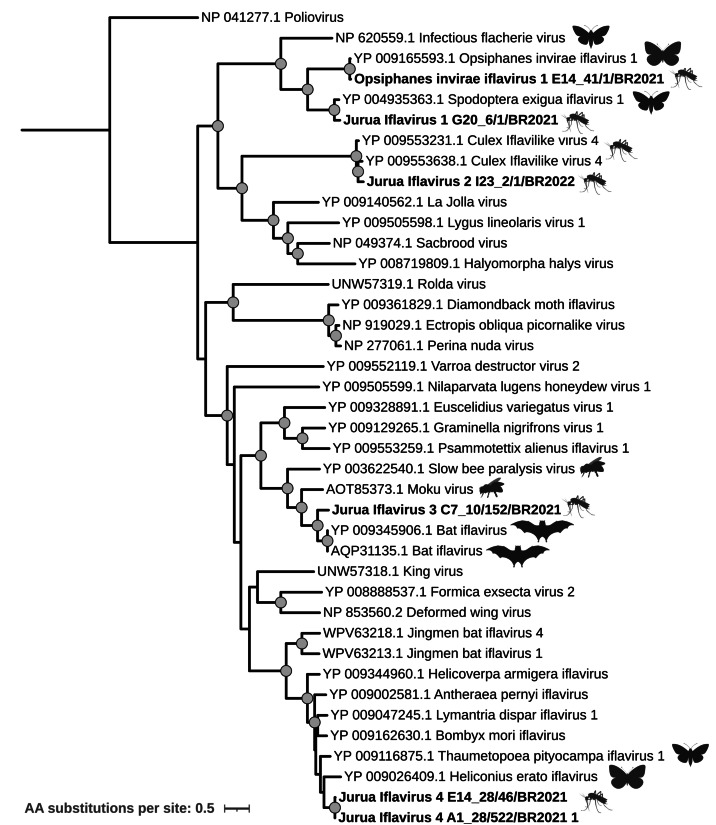 Figure 6
Figure 6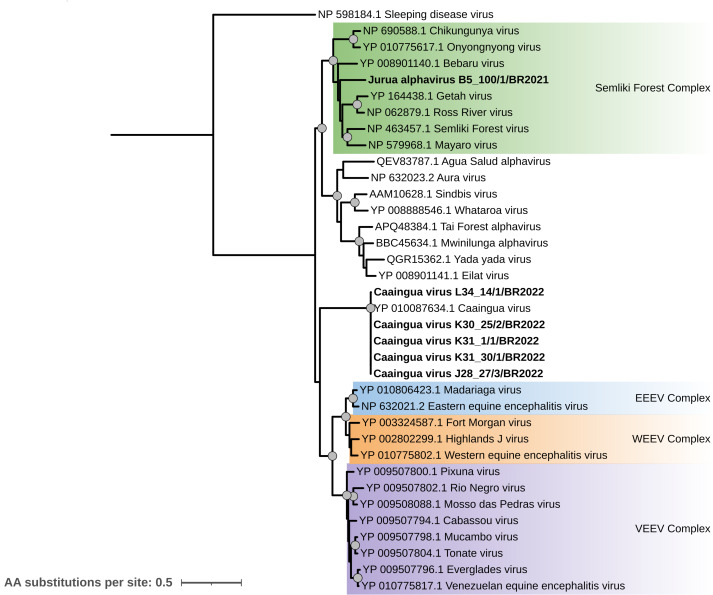 Figure 7
Figure 7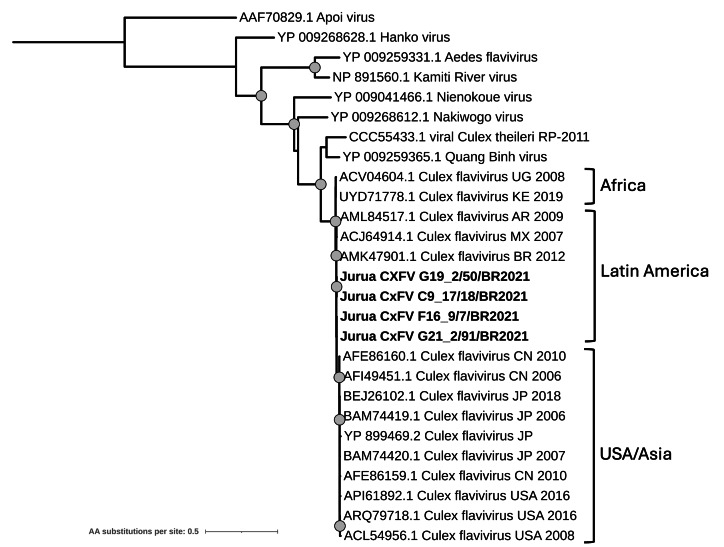 Figure 8
Figure 8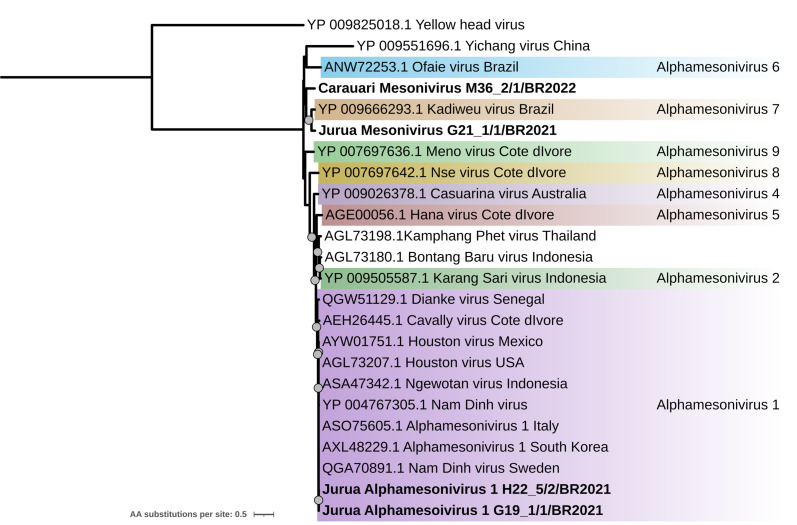 Figure 9
Figure 9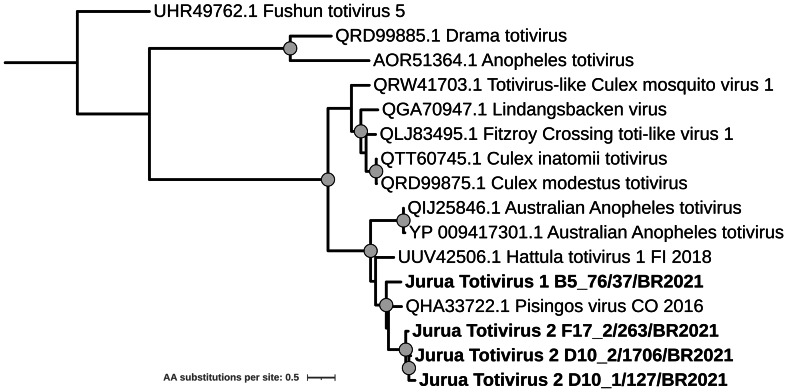 Figure 10
Figure 10- —NSF Postdoctoral Fellowship in Biology
- —Tartar Microbiology Fellowship from Oregon State University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Plant Virus Research Studies · Genomics and Phylogenetic Studies
Introduction
Arthropods play a crucial role in the transmission of pathogens, posing significant risks to both human and animal health. Among these, mosquitoes are particularly concerning as vectors for numerous pathogens and hosts of a variety of underexplored and potentially emerging viral diseases. These include arthropod-borne viruses (arboviruses), which cycle between mosquitoes and vertebrate hosts, as well as insect-specific viruses (ISVs), which are restricted to insect hosts (Bolling et al., 2015; Weaver & Reisen, 2010). RNA viruses, known for their high mutation rates and genetic diversity, are particularly prevalent among mosquito-borne viruses. Notable examples include dengue (DEN), chikungunya (CHIK), yellow fever (YF), and West Nile (WN) viruses, making arboviruses a primary focus of viral monitoring and human health research.
The high abundance and diversity of RNA viruses reported in mosquitoes support the idea that arthropods may have played a significant role in viral evolution, potentially serving as reservoirs where insect-specific viruses could evolve into dual-host viruses capable of infecting both insects and vertebrates (Öhlund, Lundén & Blomström, 2019). The ecological complexity and the interaction between these vectors and their environment create opportunities for viral diversity to emerge and persist, with direct implications for public health and conservation (Liu et al., 2023). Consequently, understanding the diversity and dynamics of RNA viruses in mosquito populations is essential for discerning viral evolution and assessing the potential for spillover events, aiding surveillance efforts, and facilitating the early detection and mitigation of emerging infectious diseases.
In recent years, the development of advanced sequencing technologies, such as high-throughput sequencing (HTS), has transformed our ability to analyze viral communities (viromes) across diverse hosts and environments (Dominguez-Huerta et al., 2023). These methodological advancements have proven crucial for examining mosquito-associated viruses and their ecological contexts, particularly in remote and lesser-studied geographical areas (Liu et al., 2023; Moonen et al., 2023). Surveillance efforts have continually expanded our understanding of viral diversity and evolution, revealing new host associations, novel viral lineages, and even new families (Bolling et al., 2015; Hermanns et al., 2023; Hollingsworth et al., 2023; Sadeghi et al., 2018). These discoveries illustrate that the tremendous diversity of viral communities in mosquitoes is just being uncovered and highlight the significance of continued research to explore this complexity and its potential impact on animal and human disease.
Biodiverse and ecologically complex regions are essential for understanding the full scope of virus-host interactions and the factors that shape viral communities. Mosquitoes in these areas interact with a diverse range of hosts and environmental conditions, facilitating viral evolution and potential cross-species transmission. Studying these ecosystems not only expands our knowledge of viral evolution and host associations but also helps establish a more complete picture of how viruses are distributed across a range of different habitats. Given the limited surveillance efforts in many of these remote regions, targeted investigations are necessary to uncover the extent of viral diversity and its broader ecological significance.
The Amazon Basin constitutes one of the most biologically rich and complex regions on Earth, harboring a vast array of ecosystems and species, including over 100 mosquito species (De Aquino et al., 2022; Garda, Da Silva & Baião, 2010; Hutchings et al., 2018; Mittermeier et al., 2003). This immense diversity presents a unique opportunity for studying these interactions as the human footprint continues to expand into these largely uninhabited regions, potentially leading to spillover events when communities increasingly interact with wildlife. In this study, we conducted a broad and deep survey of RNA viruses from mosquito populations from the Jurua River region in the Western Amazon Basin. Using HTS and de novo assembly, we analyzed 211 mosquito pools collected across a river transect of approximately 150 Kilometers in the Amazon Basin, encompassing a variety of habitats. This effort resulted in the classification of 51 viral Metagenomic Assembled Genomes (vMAGs) into 18 tentative novel viral species from a diverse range of families and 33 viral sequences as strains of nine previously described viruses. Our analysis focused on characterizing the diversity and abundance of these viruses, circulating in this ecologically rich yet understudied area. This research provides new insights into the viral diversity present in Amazonian mosquitoes, offering a broader perspective on the range of RNA viruses associated with mosquito populations in this region. By characterizing viral genomes from pooled mosquito samples, this study expands the known diversity of RNA viruses and highlights the potential ecological roles of mosquitoes in viral maintenance and transmission.
Materials and Methods
Samples collection
Mosquito sampling was conducted during two field seasons across 13 locations along the Jurua River region in the Western Amazon (Fig. 1). These locations were designated A through M, with multiple sampling sites identified within each location. Locations were sampled in alphabetical order; A to H were sampled from mid-September through October in 2021, while locations I to M were sampled from mid-September through mid-October in 2022. Each site was assigned a unique alphanumeric code to delineate specific areas (e.g., A1, A2, A3, B4, B5, B6, etc.). Additionally, multiple mosquito pools were collected at each sampling site, serving as replicates for the study (e.g A1_13, A1_28, B4_20, etc.).
Map of the region showing the sampling sites along the Jurua River.Locations are marked with different colors, corresponding to the legend on the right. The insert in the top left provides geographic context, with the study area highlighted by a green square in a zoomed-out map of South America.
To maximize mosquito capture, 12 CDC UV light traps were deployed per grid at each site for a total of 36 traps per location. Mosquitoes were trapped over two consecutive trap nights (∼48 h) and collected every 24 h, resulting in two discrete collections per trap. Following collection, mosquitoes were sorted at the trap and trap night level to isolate females, and then pooled at 50 individual females per sample and stored in RNA/DNA Shield (Zymo Research, Irvine, CA, USA) to preserve nucleic acids for sequencing. In total, we processed 211 mosquito pools from all 37 different sampling sites for RNA sequencing (Fig. 1, Table S1).
Field sample collections were conducted under permits issued by the Instituto Chico Mendes de Conservacão da Biodiversidade (ICMBio) in 2021 and by the Secretaria de Estado do Meio Ambiente do Amazonas (project authorization number 034/2022) in 2022.
Samples processing
Total nucleic acids were extracted from the mosquito pools using the Quick-DNA-RNA Pathogen Miniprep Kit (Zymo Research, Irvine, CA, USA). rRNA depletion and RNA sequencing library preparation were performed using the Illumina Stranded Total RNA Prep with Ribo-Zero Plus kit (Illumina, San Diego, CA, USA). The libraries were sequenced using two full flow cells of the Illumina NovaSeq 6000 platform at the University of Oregon’s Genomics and Cell Characterization Core Facility (GC3F). The sequencing was performed on S4 flow cells in paired-end format with a read length of 2 × 150 bp.
Data analysis
Raw reads quality was assessed with fastQC v0.11.9 (Andrews, 2010) and multiQC v1.14 (Ewels et al., 2016). Trimming of the reads was performed using fastp v0.23.2 (S. Chen et al., 2018) with the following parameters: minimum average quality of 25, minimum base quality of 20, minimum read length of 50, minimum complexity of 30%, remove poly-A, maximum of 2 Ns, auto-detect and remove adapters. Trimmed paired-end reads were subjected to a normalization step using bbnorm tool from BBTools (Bushnell, 2023), in order to reduce the number of total reads before assembling (target = 40, min = 0; all other parameters default). De novo assembly was performed using rnaSPAdes v3.15.3 (Bushmanova et al., 2019) with k-mer values 77, 99, and 127, selected to balance sensitivity and accuracy. Obtained contigs were filtered by length (minimum length of 1,000 bp) and used to perform a BLASTX search using DIAMOND v2.1.9 (Buchfink, Reuter & Drost, 2021) in sensitive mode and the latest RefSeq viral protein database (as of June 2025). A minimum bitscore of 300 and a 70% identity were used as a threshold to classify contigs as viral according to the BLASTX outputs. Viral protein sequences were obtained using Prodigal v2.6.3 (Hyatt et al., 2010) in metagenomic mode (-p meta) using NCBI standard genetic code 1 (-g 1), with default settings otherwise. CheckV (Nayfach et al., 2021) was run on viral contigs as an additional verification step to screen for potential contamination and to summarize hallmark-gene evidence alongside our Prodigal annotations. For single-contig genomes, CheckV completeness estimates were used as indicators and are provided for context (Table S2). Raw reads were mapped back to viral contigs and quantified using Kallisto v0.48.00 (Bray et al., 2016), which reports abundance as transcripts per million (TPM). TPM normalizes estimated counts by contig length and by overall sequencing depth, providing normalized measures of viral abundance.
For each putative viral contig, the family of the top hit was recorded as a candidate classification. For family-level assignment, we then built maximum-likelihood phylogenies of the RNA-dependent RNA polymerase (RdRp) or the appropriate ORF (e.g., polyprotein ORF for Iflaviridae) that included representative members of the family together with outgroup taxa from related families. Sequences that clustered with strong support within the corresponding family clade were assigned to that family. Within these family-level assignments, tentative new viruses were designated using 95% amino acid identity in the analysed protein sequence (RdRp, or the polyprotein for Iflaviridae) as a primary criterion, supported by phylogenetic analyses to confirm their evolutionary relationship. This threshold is directly in line with International Committee on Taxonomy of Viruses (ICTV) species demarcation criteria for Phasmaviridae and Phenuiviridae (L/RdRp 95% amino acid identity between species) and is more conservative than the 90% capsid protein identity used between species in Iflaviridae. For the remaining families in which we identified putatively novel viruses (e.g., Togaviridae, Mesoniviridae, Totiviridae), for which ICTV does not define a single RdRp/polyprotein cutoff, we used this 95% identity threshold as a conservative indicator of potential novelty, interpreted together with phylogenetic context. Additional factors, including associated Genbank metadata such as sampling location, collection date, and the host organism, were also considered to assess their novelty.
For contigs whose only viral matches were to poorly characterized hypothetical proteins and that lacked supported genome segments or hallmark genes, we performed additional BLAST searches against the NCBI nr database. Contigs whose best hits were to non-viral proteins (e.g., insect genes) were considered host-derived and removed from the viral dataset.
Multiple sequence alignments (MSA) were performed with mafft v7.520 (Katoh & Standley, 2013) with the–auto parameter using protein sequences from this study and reference sequences retrieved from the GenBank database. When multiple sequences were highly similar, CD-HIT v4.8.1 (Fu et al., 2012) was applied with a 98% identity threshold to retain only representative sequences. MSAs were trimmed using ClipKit v2.3.0 (Steenwyk et al., 2020) in the kpi-smart-gap mode to retain only parsimony-informative sites. Substitution model selection and phylogenetic analysis were performed using IQ-TREE 2 (Minh et al., 2020) applying 1,000 bootstrap replicates (-b 1000) and 1,000 replicates for the approximate likelihood ratio test (-alrt 1000). Phylogenetic trees were visualized and edited using iTOL v6 (Letunic & Bork, 2024).
Results
Mosquito capture, sorting, and pooling reveals a diverse population of RNA viral Metagenomic Assembled Genomes (vMAGs)
A total of 10,550 mosquitoes were collected from 13 locations across the Jurua river region in the Western Amazon, from which 211 pools of fifty putative female mosquitoes were created, resulting in 211 RNAseq libraries (one of the largest datasets of its kind, NCBI BioProject PRJNA1223432). High-throughput sequencing generated approximately 4.3 TB of sequencing data, covering a ∼150-kilometer transect of the Jurua River (Fig. 1). This dataset provided a comprehensive foundation for identifying and characterizing the RNA viral diversity associated with mosquitoes in this ecologically complex and understudied region.
Mosquito RNAseq libraries had an average of 26,597,204 high-quality paired reads per library (Table S3). Following de novo assembly, 209 libraries contained contigs longer than 1 Kb, with 138 of these containing at least one >1 Kb contig classified as viral based on conservative DIAMOND and CheckV results, as described in methods. While all the locations (A to M) covered in this study remained represented in the final 138 libraries used for viral population analyses, the number of total sampling sites was reduced from 37 to 35, as libraries from two sampling sites (K_29 and L_33) did not contain sufficiently high-quality viral contigs.
BLASTX results identified 650 viral contigs longer than 1 Kb (based on parameters described in the methods), representing 18 different viral RNA families (Fig. 2). The most prevalent viral family was Phasmaviridae, identified in over 40% of the pools. Rhabdoviridae and Flaviviridae were found in 10–20% of the pools, while families Phenuiviridae, Iflaviridae, and Togaviridae were detected in 5–10%. CheckV results indicated that 51 viral Metagenomic Assembled Genomes (vMAGs) had a completeness level greater than 95%, with thirty-four showing 100% completeness.
Overall virus diversity and abundance .The bar chart on the left shows the prevalence of viral families identified in this study across the different pools (libraries). The heatmap on the right illustrates their respective abundances (represented as Log(TPM+1)) across the sampling sites.
Within the identified viral families, 25 contigs were classified as belonging to the family Retroviridae. However, due to the challenges in distinguishing between endogenous and exogenous retroviruses in this type of sequencing data, these sequences were excluded from further analysis and are not included in the results presented here.
Genetic characterization of viral sequences reveals unknown diversity of viral families in the Amazonas Basin
Protein sequences derived from the vMAGs were used for phylogenetic analyses together with representative reference sequences to compare our viruses with previously reported strains and to resolve within-family diversity (see Methods). Based on the criteria described above, a total of 82 viral strains were classified as either novel viruses or strains from previously reported viruses (Table 1). This section presents the results for viral families identified, focusing on sequences that were sufficiently long and overlapping to allow for MSA and phylogenetic analyses, enabling their assignment to a viral taxon.
Table 1: Viral strains obtained in this study that were classified at a species level.Corresponding families, pools from which they were obtained, strain names, and GenBank accession numbers.
Phasmaviridae
We identified ninety-eight contigs associated with the Phasmaviridae family from forty-two pools across sixteen different sampling sites. Members of this family are characterized by a segmented genome with three negative-sense RNA segments (L, M, and S). BLASTX analysis revealed best hits to Jonchet virus (one sequence), Anopheles orthophasmavirus (one sequence), Wuhan mosquito virus 1 (one sequence), and Coredo virus (ninety-five sequences). Among these, seventy-three sequences corresponded to the L segment, from which thirty-nine RdRp sequences were used to construct an MSA alongside representative sequences from all known Phasmaviridae genera.
Phylogenetic analysis showed that the sequence obtained from pool A3_2 (strain A3_2/780/BR2021) clustered with other strains from the genus Jonvirus (Spilikin, Jonchet, and Mikado virus), forming a monophyletic clade with high statistical support (Fig. 3). The amino acid sequence identity between this strain and previously reported Jonvirus was 71.2%, 71.5%, and 72.0% with Mikado, Spilikins, and Jonchet viruses, respectively. We tentatively named this virus Jurua Jon-like virus.
Maximum likelihood phylogenetic tree based on the L segment (2,066 aa sites) of the Phasmaviridae family.Sequences obtained in this study are shown in bold. Circles on nodes indicate bootstrap values ≥85, and colors represent the different genera within this family. Numbers within parentheses indicate the number of sequences represented by a single tip after clustering analysis using CD-HIT with a 98% aa identity threshold.
The RdRp sequence obtained from pool B6_22 (strain B6_22/775/BR2021) shared 97% amino acid identity with Anopheles triannulatus orthophasmavirus, indicating that it corresponds to a variant of this virus. The sequence obtained from pool J27_25 (strain J27_25/5/BR2022) showed 76% identity with Wuhan mosquito virus 1. We named this tentative new virus Jurua orthophasmavirus. Phylogenetic analysis grouped these viral strains in a monophyletic clade within the genus Orthophasmavirus with high statistical support, alongside Anopheles stephensi orthophasmavirus (Fig. 3).
The thirty-six sequences closely related to Coredo virus were grouped into eleven clusters using CD-HIT analysis with a 98% amino acid identity threshold (Table S4). For better visualization, only representative sequences from each cluster were employed in the phylogenetic analysis. These sequences formed a major clade composed of Coredo and Coredo-related sequences. Using a 95% amino acid identity threshold, the sequences were further classified into four novel clades named Jurua Coredo-like virus 1–3 (Fig. 3; Table S4).
Phenuiviridae
Thirteen contigs from seven pools across six sampling sites were identified as belonging to the Phenuiviridae family. The most closely related known viral species were Narangue virus (five pools) and Cumuto virus (two pools). Five RdRp sequences were included in a phylogenetic analysis alongside representative sequences from the family (Fig. 4). The resulting phylogenetic tree revealed multiple clades, reflecting the family’s diversity and its classification into several genera.
Maximum likelihood phylogenetic tree for the L segment (2,509 amino acid sites) from the Phenuiviridae family.Sequences obtained in this study are shown in bold. Circles on nodes indicate bootstrap values ≥85. Colors represent the different genera within this family.
Three viral sequences grouped within the genus Mobuvirus with high statistical support, representing two tentatively novel viruses closely related to Narangue virus: Jurua Narangue-like virus (strain B4_1/182/BR2021, 84% RdRp amino acid identity) and Jurua mobuvirus (strains C7_2/2/BR2021 and D10_2/25/BR2021, 73.3% and 71.4% RdRp amino acid identity, respectively).
Sequences from pools E14_28 and B6_22, closely related to Cumuto virus, clustered within the genus Goukovirus with high statistical support. These sequences correspond to two tentative new viruses: Jurua goukovirus and Jurua Cumuto-like virus, obtained from pools E14_28 and B6_22, respectively (Fig. 4). While both viruses had Cumuto virus as their best BLASTX hit, they showed distinct amino acid identities with their RdRp protein sequence: 74.7% for Jurua goukovirus and 83.5% for Jurua Cumuto-like virus.
Peribunyaviridae
All three genomic segments of a virus from the genus Orthobunyavirus were identified in pool F17_5. The segments were 6,934 bp (L segment), 5,044 bp (M segment), and 1,315 bp (S segment) in length. BLASTX analysis revealed amino acid identities of 98.1%, 83.7%, and 97.1% for the L, M, and S segments, respectively, compared to Alajuela virus. Based on the established 95% species threshold for the RdRp (L segment) and phylogenetic analysis, these results indicate that the L segment corresponds to a strain of previously described Alajuela virus reported in Argentina (Nunes et al., 2014).
Nairoviridae
Twelve contigs from twelve different mosquito pools were associated with the Nairoviridae family. For all these contigs, the best match was to a hypothetical protein encoded by the M segment of Blattodean nairo-related virus (also known as red goblin roach virus 1), with amino acid identities ranging from 72% to 75%. According to the ICTV, this hypothetical protein is characteristic of the genus Ocetevirus, the only genus within the Nairoviridae family where this protein has been reported. The identified contigs ranged in length from 1,035 bp to 1,775 bp. Notably, no contigs were associated with the L segment (encoding the RdRp protein) or the S segment (encoding the nucleocapsid protein) of the Nairoviridae genomes. Additionally, none of the contigs matched the Glycoprotein precursor, the other gene encoded by the M segment.
To evaluate alternative explanations, we further searched these twelve contigs against the NCBI nr database using BLASTN. In all cases, the best-scoring hit was a mosquito phosphatase gene (accession XM_038252539.1) showing nucleotide identity values greater than 93%. These results indicate that the contigs are most likely host-derived sequences rather than actual Nairoviridae segments. We therefore no longer treat them as evidence of Nairoviridae presence in our pools and have removed them from our viral catalog and all downstream analyses.
Rhabdoviridae
Forty-six contigs from sixteen pools were identified as belonging to the Rhabdoviridae family, with Merida virus (MERDV) as their best hit according to BLASTX results. Nine corresponded to full vMAGs (∼11 Kb), from which we could annotate the five genes that compose the genome and encode for the nucleoprotein, phosphoprotein, matrix protein, glycoprotein, and RdRp. Full-length RdRp protein sequences were employed for the genetic characterization of the strains. All sixteen RdRp sequences obtained from the complete and nearly complete genomes were highly similar, forming a single cluster with 98% amino acid identity threshold based on CD-HIT analysis (Fig. S1). Strain C7_13/BR2021 from pool C7_13 was employed as a representative sequence of this cluster for better visualization of the tree.
Amino acid identities among the RdRp sequences showed that Merida virus sequences from this study present high amino acid identity (≥98%) with other Merida virus sequences from the Americas (USA and Mexico). In contrast, they exhibited slightly lower identity values (95–96%) with sequences from Europe (e.g., Sweden, Georgia, and Turkey) and Asia (e.g., China) (Fig. 5; Fig. S1).
Maximum likelihood phylogenetic tree for the RdRp protein sequence (1,202 sites) from the Merhavirus genus.Sequences obtained in this study are shown in bold. Circles on nodes indicate bootstrap values ≥85. Numbers within parentheses indicate the number of sequences represented by a single tip after clustering analysis using CD-HIT with a 98% aa identity threshold. Geographic locations for the Merida virus strains are indicated.
Xinmoviridae
Two complete vMAGs were recovered from pools M36_2 and M37_4, both classified within the genus Madalivirus, which currently includes only two reported species: Anopheles darlingui virus and Anopheles marajoara virus. The recovered genomes correspond to strains of Anopheles darlingi virus, originally isolated in 2015 in Brazil (Scarpassa et al., 2019), with RdRp amino acid identities of 99.8% and 99.9% for pools M36_2 and M37_4, respectively. When compared to all available Madalivirus strains, the RdRp sequences of our genomes showed identity values greater than 98% with anopheles darlingi virus strains and greater than 70% with Anopheles marajoara virus strains (Fig. S2). Additionally, we were able to annotate all five previously reported Madalivirus genes in both genomes: nucleoprotein, transmembrane protein, glycoprotein 1, glycoprotein 2, and RdRp.
Iflaviridae
A total of eleven contigs from seven different pools were identified as belonging to the Iflaviridae family. Two complete vMAGs, each corresponding to a single polyprotein ORF, were recovered from pools E14_41 (strain E14_41/1/BR2021, 10,155 bp) and G20_6 (strain G20_6/1/BR2021, 10,239 bp). Strain E14/1/BR2021 was classified as Opsiphanes invirae iflavirus, with 97.0% amino acid identity across the polyprotein. In contrast, strain G20_6/1/BR2021 shared 83.4% amino acid identity with its best match (Spodoptera exigua iflavirus) and was therefore tentatively named Jurua iflavirus 1. Phylogenetic analysis using representative sequences from the Iflaviridae family grouped these genomes within a well-supported monophyletic clade that also included infectious flacherie virus (Fig. 6). Except for the sequences obtained in this study (associated with mosquito hosts), all the previously reported viral strains from this clade were associated with lepidopteran hosts.
Maximum likelihood phylogeny of a partial (2,066 sites) iflavirus polyprotein sequence.Sequences obtained in this study are identified in bold. Small icons represent the known hosts for the closest related viruses to the sequences obtained in this study. Bootstrap values greater than 85 are shown with circles.
In addition to the complete genomes, four partial polyprotein sequences were also included in the phylogenetic analysis. One of them, recovered from pool I23_2 (strain I23_2/1/BR2022), shared 87.2% amino acid identity with culex iflavilike virus 4 (Fig. 6). Although closely related, the level of divergence observed at the polyprotein level suggests that this strain represents a distinct virus, which we tentatively named Jurua iflavirus 2.
Another partial polyprotein sequence, obtained from pool C7_10, showed 74.2% amino acid identity to two Bat iflavirus genomes and clustered in a clade that also includes Moku virus and Slow bee paralysis virus, both originally isolated from hymenopteran hosts (Fig. 6). This virus was named Jurua iflavirus 3.
Sequences from pools E14_28 and A1_28 grouped closely, with 90.1% amino acid similarity between them, and they both had Heliconius erato iflavirus as their best hit, showing 78.2% and 79.1% amino acid identity, respectively. These two sequences represent strains from the same virus that we tentatively named Jurua Iflavirus 4. The clade where these strains are grouped is composed of viral strains isolated from lepidopteran hosts (Fig. 6).
Togaviridae
Thirteen contigs from six different pools were identified as belonging to the family Togaviridae. Five complete vMAGs and one complete nsp4 protein sequence were obtained (Table 1) and used to perform a phylogenetic tree with representative sequences of the family. Five of these sequences (from pools L34_14, K30_25, K31_1, K31_30, and J28_27) correspond to strains of the previously reported Caaingua virus (≥98% RdRp amino acid identity), isolated in Southern Brazil in 2017 (Tschá et al., 2019). The Caaingua virus strains form a monophyletic clade with high statistical support, and do not cluster with any of the previously described complexes for this family (Fig. 7).
Maximum likelihood phylogenetic tree for the RdRp protein sequence from the Togaviridae family.Sequences obtained in this study are shown in bold. Circles on nodes indicate bootstrap values ≥85. Colors represent previously described major clades identified for this family: Western Equine Encephalitis virus (WEEV), Semliki Forest virus, Eastern Equine Encephalitis virus (EEEV), and Venezuelan Equine Encephalitis virus (VEEV) Complexes.
The remaining vMAG for this family (from pool B5_100) clusters within the Semliki Forest Complex with high bootstrap values, grouping with well-known arboviruses like Chikungunya, Getah, Ross River, Semliki Forest, and Mayaro viruses (Fig. 7). Based on amino acid identity, the closest strain for this genome was Semliki Forest virus, showing 70.9% amino acid identity with the RdRp protein sequence. Thus, this genome was classified as a novel virus tentatively named Jurua alphavirus.
Flaviviridae
A total of thirty-three contigs from eleven mosquito pools were associated with the Flaviviridae family, all showing more than 90% amino acid identity with the RdRp protein of Culex flavivirus (CxFV) (Fig. S3). Phylogenetic analysis was conducted using sequences from three pools, which grouped with previously reported CxFV strains isolated in Latin America, including one strain collected in Brazil in 2012 (Fig. 8). The phylogenetic tree structure reflects a geographic pattern, clustering the different CxFV strains into three main clades: Africa, Latin America, and a group containing sequences from USA and Asia. This aligns with the genotypic classifications described in previous studies (Bittar et al., 2016; Miranda et al., 2019).
Maximum likelihood phylogenetic tree for the RdRp protein sequence from Culex flavivirus and closely related species.Sequences obtained in this study are shown in bold. Circles on nodes indicate bootstrap values ≥85. Geographic locations for the CxFV strains are indicated.
Mesoniviridae
Eleven contigs from seven different pools were identified as belonging to the Mesoniviridae family. Three contigs corresponded to complete vMAGs (pools G19_1, G21_1, and M36_2). Partial RdRp sequences for four strains were employed to construct a phylogenetic tree using previously reported Alphamesonivirus viral strain sequences. The phylogenetic tree placed strains M36_2/1/BR2021 and G21_1/1/BR2021 within branches closely related to Kadiweu virus (isolated in Brazil in 2010 and also called Alphamesonivirus 7 according to Newton et al. (2020), with 75.3% and 87.6% identity, respectively. These two tentative novel viruses were named Carauari mesonivirus (strain M36_2/1/BR2021) and Jurua mesonivirus (strain G21_1/1/BR2021) (Fig. 9).
Maximum likelihood phylogeny of the complete Mesonivirus ORF1b protein sequence.Sequences obtained in this study are identified in bold. Bootstrap values greater than 85 are shown.
The other two strains obtained for this family, H22_5/2/BR2021 and G19_1/1/BR2021, presented identical amino acid sequences and were grouped within a clade composed of strains of Alphamesonivirus 1 which have been isolated worldwide (Fig. 9). Amino acid identity values between all the sequences from the Mesoniviridae dataset are found in Fig. S4.
Totiviridae
Five contigs from five different pools were associated with the Totiviridae family, all of them with Australian Anopheles totivirus as the best hit according to DIAMOND results. Four RdRp protein sequence were retrieved from the contigs sequences and used to generate a phylogenetic tree with Totivirus strains isolated from mosquito species retrieved from the GenBank database. Phylogenetic tree grouped the sequences herein obtained and Australian Anopheles virus sequences together in a clade with big statistical support, along with Hattula virus 1 and Pisingos virus, isolated from Finland and Colombia, respectively (Fig. 10). Phylogenetic tree, as well as amino acid distance analysis, showed that the sequences obtained in this study correspond to two tentative novel viruses named Jurua totivirus 1–2 and closely related to Pisingos virus (Fig. 10, Fig. S5).
Maximum likelihood phylogeny based on the partial (553 sites) RdRp protein sequence of Totivirus strains isolated from mosquitoes.Sequences obtained in this study are identified in bold. Bootstrap values greater than 85 are indicated with a circle.
Discussion
This study provides a comprehensive analysis of the RNA viral communities associated with wild-caught adult female mosquito populations from the Jurua River region in the Amazon Basin. Leveraging one of the largest mosquito pool datasets to date, we identified RNA viruses from eighteen families, including twenty-one complete or nearly complete viral metagenomic assembled genomes (vMAGs) with high completeness (≥95% completeness according to CheckV) and remarkable diversity. Our pooling strategy prioritized broad geographic coverage and viral discovery across sites, enabling the detection and genetic characterization of multiple viral taxa circulating in this understudied region. However, because pools combine multiple, often mixed-species individuals, our approach cannot resolve which viruses co-occur within individual mosquitoes or specific host species. Despite this limitation, these findings represent a significant contribution to understanding the genetic diversity of RNA viruses circulating in this biodiverse and understudied region, while also providing insights into viral evolution and ecological dynamics.
At a genome composition level, previous virome survey studies have stated that positive-sense single-stranded RNA families such as Flaviviridae, Mesoniviridae, and Iflaviridae are among the most abundant and widespread viruses in mosquitoes (Chen et al., 2024; Moonen et al., 2023; Morel et al., 2024). While these families are not the most prevalent or abundant in our samples, multiple species of known and tentatively novel viruses belonging to these families were identified in our data, consistent with earlier work. At the same time, we found that negative-sense RNA families make a particularly strong contribution to viral diversity in our system. Phasmaviridae and Phenuiviridae are both highly prevalent across pools and together account for a large fraction of the viral species recovered. This pattern suggests that in the Amazonian rainforest mosquitoes, (-)ssRNA viruses can be major contributors to species richness alongside the positive-sense families.
The contrast with many published studies, where (+)ssRNA viruses are often reported as dominant, can be explained by factors related to differences in sampling (e.g., ecosystem, host species). Most virome work to date has focused on recognized vector genera such Aedes and Culex and on sites with substantial human activity, frequently as part of arbovirus surveillance programs in urban or peridomestic environments (Hollingsworth et al., 2023; Liu et al., 2023; Moonen et al., 2023). Because mosquito virome composition is strongly structured by host species (Liu et al., 2023; Thongsripong et al., 2021; Wu et al., 2023), the predominance of (+)ssRNA families in these studies may partly reflect the ecology of a small set of well-studied vector species. In contrast, our study targets mixed mosquito assemblages from remote Amazonian rainforest, where mosquito diversity and viromes remain poorly characterized.
Moreover, a recent study on sylvatic mosquitoes in Brazil supports this view. Metatranscriptomic surveys of forest-dwelling mosquitoes from Northeast Brazil and the Cerrado biome revealed highly diverse viromes with a more balanced composition between positive and negative sense RNA viruses (Da Silva et al., 2024). Viral species belonging to families like Phasmaviridae (the most prevalent family observed in this study) and Phenuiviridae were described as part of the main components of the virome in species like Coquillettidia hermanoi and Limatus durhamii, which are rarely included in urban mosquito virome work. Together with our findings, these results suggest that (-)ssRNA viruses can be major contributors to diversity in sylvatic mosquito communities, even though they are less visible in datasets dominated by urban vector species.
Expanding the knowledge of Bunyaviricetes in Amazonia
The recently elevated class Bunyaviricetes includes hundreds of segmented RNA viruses, many of which are pathogenic to humans, animals, plants, and fungi (Kuhn et al., 2024). In this study, we classified vMAGs into three families within this class: Phasmaviridae, Phenuiviridae, and Perybunyaviridae. Across the analyzed pools, Phasmaviridae emerged as the most prevalent family, represented by members of the genera Orthophasmavirus and Jonvirus. Within the Orthophasmavirus genus, a substantial number of vMAGs were closely related to Coredo virus, represented by one strain isolated from Mansonia titillans mosquitoes in Colombia in 2016. Using the ICTV-defined threshold of 95% amino acid identity in the RdRp amino acid sequence for species differentiation, our results suggest the presence of a highly diverse array of Coredo-like viruses in the Jurua region, likely representing multiple unreported species sharing a common ancestor (Fig. 3). These results support the need for continued study of this viral group, which appears to be an integral part of the local mosquito virome.
In addition to Coredo-like viruses, two strains within the Orthophasmavirus genus were identified: strains B6_22/775/BR2021 and J27_25/5/BR2022. These sequences form a well-supported monophyletic clade with other viruses isolated from Anopheles mosquitoes, including Anopheles triannulatus orthophasmavirus, previously reported in the Amazon (Scarpassa et al., 2019), and Anopheles stephensis orthophasmavirus, reported in Japan (Matsumura et al., 2024). While we cannot confirm the specific mosquito hosts of the newly identified viral strains, the clustering of these sequences with previously reported viruses associated with Anopheles species suggests the potential for similar host associations.
Further, our discovery of a new Jonvirus strain marks the first report of this genus outside Africa (Hermanns et al., 2023; Marklewitz et al., 2015). This finding suggests a broader geographical distribution for Jonvirus and underscores the utility of HTS for uncovering the evolutionary relationships and diversity of the Phasmaviridae family.
The detection of viral strains from the Phenuiviridae family revealed representatives from the genera Mobuvirus and Goukovirus (Fig. 4). These genera consist of insect-specific viruses (Sasaya et al., 2023), and the sequences identified in this study represent two tentative novel viruses closely related to Narangue virus and Cumuto virus, previously isolated from Mansonia and Culex mosquitoes, respectively (Auguste et al., 2014). The distribution of these viruses in tropical regions such as Colombia and Trinidad aligns with the environmental characteristics of the Amazon, highlighting the region’s unexplored viral diversity and the need for further exploration.
A putative complete set of genomic segments from the Perybunyaviridae family was obtained from pool F17_5 and assigned to the Gamboa virus complex within the genus Orthobunyavirus. The L segment showed sequence identity levels to Alajuela virus above the species demarcation criteria from ICTV, indicating the presence of this species in the pool. While the M and S segments also had Alajuela virus as their best BLASTX hit, these proteins are less conserved and no formal ICTV thresholds are defined for them. Because these data derived from pooled mosquitoes, we cannot exclude the possibility that the three segments originate from more than one closely related Gamboa complex virus (for example through reassorment or co-infection). We therefore report the presence of Alajuela virus based on the L segment and interpret the associated M and S segments as most closely related to Alajuela virus.
Members of the Gamboa serogroup, including Alajuela virus, have been isolated mainly from the avian-feeding mosquito Aedeomyia squamipennis across several Neotropical countries like Panama, Ecuador, Argentina, Suriname, and Brazil (Eastwood et al., 2016; Nunes et al., 2014). Transovarial transmission of Gamboa virus in A. squamipennis has also been demonstrated, indicating that this mosquito species can act as a maintenance host for Gamboa-serogroup viruses in nature (Dutary et al., 1989). While not linked to human disease, monitoring the genetic diversity of such viruses remains critical, as recombination events in Orthobunyavirus strains have led to outbreaks of pathogenic viruses, such as the recent Oropouche virus outbreak in South America (Zhang et al., 2024).
Although evidence of Nairoviridae was found (another family belonging to the Bunyaviricetes order), all identified contigs matched a single hypothetical protein unique to the genus Ocetevirus (Käfer et al., 2019) and lacked the hallmark RdRp ORF expected in this family. These contigs were later resolved as mosquito genes once we expanded the search to broader databases, leading us to remove these contigs from the viral catalog. Our re-evaluation of the putative Nairoviridae signal illustrates how easily host-derived sequences can masquerade as viral when annotations rely heavily on poorly characterized “hypothetical proteins”. Beyond the specific case of Nairoviridae, this highlights how gaps and misannotations in both viral and host genome resources can distort perceived community composition if not carefully checked. More generally, it underscores the need for conservative, multi-step annotation strategies, combining viral-focused databases with comprehensive resources such as nr, considering genome architecture and hallmark genes, and treating isolated hits to hypothetical proteins with caution, to avoid overinterpreting spurious signals in metagenomic virome studies.
Insights into Mononegavirales diversity
Within the Mononegavirales order, two families were identified: Rhabdoviridae and Xinmoviridae. Rhabdoviridae was the second most prevalent family, with sixteen new vMAGs corresponding to Merida virus. This virus was originally reported in Mexico in 2016 (Charles et al., 2016) and later identified in the USA (Sadeghi et al., 2018). To date, Merida virus has been reported in mosquitoes from four continents and in multiple mosquito genera, including Culex, Aedes, and Haemagogus (Moonen et al., 2023). Our data extends its known distribution to the Western Amazon, representing the first report of Merida virus in South America. The high prevalence of Merida virus in our pools is consistent with previous surveillance studies in which rhabdoviruses, including Merida-like viruses, are among the most frequently detected and widespread mosquito-associated viruses (Moonen et al., 2023; Morel et al., 2024). While Merida virus had not been reported in South America before this study, sequences with lower amino acid identity values (51%) to Merida virus were previously reported in the Amazon region (Da Silva Neves et al., 2021), suggesting the circulation of unreported closely related species in the region that warrants further investigation.
Phylogenetic analysis revealed three distinct clades, likely reflecting the geographic separation between strains from Asia, Europe, and the Americas. For generalist mosquito-associated viruses such as Merida virus, the ability to infect multiple mosquito species has been proposed as one explanation for their global distribution, along with co-radiation with culicids during their evolution and spread (Morel et al., 2024). Furthermore, other genera within Rhabdoviridae, such as Nucleorhabdovirus and Cythorhabdovirus, are known to infect plants and animals while using arthropods as vectors, facilitating the spread of these viruses (Whitfield et al., 2018) and demonstrating that multi-host life cycles can occur within this family. Whether Merida virus and related mosquito-associated rhabdoviruses share such complex ecologies is currently unclear, and more comprehensive screening of mosquitoes and potential non-mosquito hosts will be needed to clarify their evolution and host range.
Iflaviridae: host associations and ecological links
The Iflaviridae family comprises viruses with a non-segmented, (+)ssRNA genome. To date, and according to the ICTV, Iflaviruses exclusively infect arthropod hosts, with the majority reported in insects. While this family currently lacks formal genus-level classifications, phylogenetic analyses have consistently revealed the presence of distinct clades with strong statistical support, suggesting shared ancestry among viral strains (Valles et al., 2017).
The viral sequences identified in this study fall into four of these distinct clades and are closely related to viruses previously isolated from various insect orders, including Diptera (e.g., flies) and Lepidoptera (e.g., moths and butterflies). This is consistent with earlier studies indicating that Iflaviridae evolution does not follow a strict host-based pattern, as each clade contains viruses infecting hosts from different insect groups (Valles et al., 2017).
A particularly notable result is the close relationship between one of our sequences (a tentatively new virus named Jurua iflavirus 3) and Bat iflavirus (Fig. 6). Viral strains from the Iflaviridae family, such as Bat iflavirus and King virus, have been associated with bats (He et al., 2013; Juergens, Huckabee & Greninger, 2022; Yinda et al., 2017). However, these strains have been isolated from either bat guano or intestinal contents, making it difficult to distinguish between true vertebrate infection and viruses derived from ingested arthropods. Thus, the most plausible explanation is that Jurua iflavirus 3 represents an ISV that can also be detected in bat-associated samples via dietary or environmental exposure, rather than an arbovirus lifestyle. Our phylogenetic results therefore point to ecological links between mosquitoes and insectivorous vertebrates. Further studies are needed in order to determine whether any iflaviruses are capable of replicating in vertebrate hosts.
Togaviridae and Flaviviridae: implications for public health and virus evolution
The Togaviridae family consists of non-segmented, (+)ssRNA viruses currently classified into a single genus, Alphavirus (Strauss & Strauss, 1994). In this study, we identified and characterized the complete genome of a novel alphavirus, Jurua alphavirus (11Kb genome length), which was classified within the Semliki Forest Complex (Fig. 7), a major clade recognized by the ICTV. Notably, insect-specific alphaviruses included in the phylogenetic analysis like Eilat virus, Aqua salad alphavirus, and Yada Yada virus, form a monophyletic clade with high bootstrap values, supporting the evidence that Jurua alphavirus is more evolutionary related to the arboviruses from the Semliki Forest Complex rather than to insect-specific viruses from the Togaviridae family. The Semliki Forest Complex includes Old-World alphaviruses known to cause diseases in humans and animals, such as Chikungunya virus and Mayaro virus. These viruses are associated with similar symptoms like fever, headache, and arthralgia (Henss et al., 2019). While Brazil is one of the countries with the highest recency and frequency of Chikungunya cases (Grabenstein & Tomar, 2023), epidemics of Mayaro fever have been reported in several regions of Latin America, including the Amazon Basin (Henss et al., 2019; Mourão et al., 2012). The identification of Jurua alphavirus, a previously unrecognized virus within this medically significant clade, underscores the need for further investigation into its pathogenic potential, host range, and prevalence in the region. Given the history of emerging alphaviruses causing epidemic outbreaks in the region (Azar et al., 2020; Resck et al., 2024), enhanced surveillance efforts are warranted to monitor the presence and possible spillover risks associated with this virus.
Additionally, this study provides four complete genomes of Caaingua virus (CAAV), an insect-specific alphavirus isolated from mosquito pools of Culex sp. in Southern Brazil in 2019 (Tschá et al., 2019). Phylogenetic analysis indicates that CAAV strains form a monophyletic clade that is basal to the New World encephalitic alphavirus side (WEE/VEE complexes, see Fig. 7) without clustering with any currently defined complex. This topology is consistent with broader alphavirus phylogenies in more recent work (Silva et al., 2022). These findings highlight the unexplored genetic diversity within the Togaviridae family and suggest that CAAV-like viruses may present a distinct evolutionary history from the better-studied Old World and New World Alphavirus complexes*.* Our recovery of multiple CAAV strains from Amazonian mosquito pools extends the known geographic and ecological range of this lineage and underscores that much of the basal diversity of Alphaviruses remains unsampled in sylvatic mosquito communities.
The Flaviviridae family, another medically significant group of (+)ssRNA viruses, includes both medically important arboviruses and Insect-Specific Flaviviruses (ISFV), which have gained particular interest for their potential interactions with pathogenic flaviviruses (Vasilakis & Tesh, 2015). Among the flaviviruses of greatest public health concern, Dengue virus, Yellow Fever virus, and Zika virus are all endemic to the Amazon region (Barreto et al., 2020; Figueiredo, 2000; Rincón et al., 2024). However, in addition to these arboviruses, ISFVs are increasingly recognized as key components of mosquito viromes, as their widespread distribution and host-restricted replication suggest they may influence the transmission dynamics of pathogenic flaviviruses (Kent, Crabtree & Miller, 2010; Talavera et al., 2018). In this study, we identified sequences corresponding to Culex Flavivirus (CxFV), an ISFV distributed worldwide.
Our findings further demonstrate the presence and repeated detection of ISFV in Amazonian mosquito populations, with sequences identified in eleven mosquito pools. The four CxFV strains characterized in this study contribute to a better understanding of the genetic diversity and geographic distribution of ISFVs. Additionally, our phylogenetic analysis supports the previously proposed genotypic classification of CxFV, which appears to follow a geographic clustering pattern (Miranda et al., 2019). Given the potential role of ISFVs in influencing the replication and transmission of medically important flaviviruses, these findings emphasize the importance of continued surveillance in the Amazon Basin to monitor co-circulating arboviruses and ISFVs and to assess their ecological interactions.
Mesoniviridae: expanding the known genetic diversity of an emerging insect-specific family
The Mesoniviridae family, classified within the Nidovirales order, consists of (+)ssRNA viruses and represents the first family in the order reported to infect insects (Zirkel et al., 2013). This makes Mesoniviridae particularly relevant from an evolutionary perspective, as its origins may provide insights into the broader evolutionary history of Nidovirales, which includes viral families of significant concern such as Coronaviridae. Mesoniviridae has been described as a missing link in the transition from small to large nidoviruses, representing an intermediate step in the genome expansion and complexity within the order (Nga et al., 2011). The study of this family is therefore critical to understanding the evolutionary pressures and mechanisms that may have led to the emergence of larger nidoviruses, including coronaviruses.
Since it was first described, the Mesoniviridae family has rapidly expanded, with new species being identified and classification systems continuously refined (Morais et al., 2022). In this study, we identified four new complete mesonivirus genomes (∼20Kb), two of which correspond to strains of Alphamesonivirus 1, the most frequently isolated mesonivirus to date. Additionally, we describe two novel mesoniviruses, Carauari mesonivirus and Jurua mesonivirus, both closely related to Kadiweu virus (KADV), also named Alphamesonivirus 7, originally isolated from the High Pantanal region in Brazil (Maia et al., 2019). These findings expand the known diversity of mesoniviruses in Brazil (which also includes Ofaie virus), demonstrating the presence of a rich but largely unexplored mesonivirus diversity in South America.
Totiviridae: emerging diversity and potential role in mosquito viromes
The Totiviridae family comprises dsRNA viruses originally known to infect fungi, protozoa, and plants (Ghabrial & Nibert, 2009). However, an increasing number of totiviruses have also been identified in insects, including mosquitoes (Fauver et al., 2016; Truong Nguyen et al., 2022; Zhai et al., 2010). Despite their widespread detection across diverse species and global distribution, mosquito-associated totiviruses remain unclassified by the ICTV, so their taxonomic placement remains incomplete. Moreover, the majority of them are known only from metagenomic data, leaving their ecology and host interactions poorly understood.
In this study, we identified two novel mosquito-associated totiviruses, Jurua totivirus 1 and Jurua totivirus 2, which are closely related to Pisingus virus, a totivirus isolated from Psorophora funiculus in Colombia in 2016. Our findings add to the growing catalog of mosquito-associated totiviruses and extend the known geographic range of this lineage into the Western Amazon. This pattern is consistent with surveillance studies that report frequent detection and high prevalence of totiviruses in mosquito populations (Coatsworth et al., 2022; Truong Nguyen et al., 2022). However, further analyses will be needed to clarify their host range and potential ecological roles in mosquito biology and virus-host interactions.
Conclusions
This study represents one of the most extensive viral surveillance efforts conducted in the Amazon Basin to date, analyzing over 200 mosquito pools across a vast and biodiverse region. By leveraging HTS and de novo assembly, we uncovered an unprecedented diversity of RNA viruses, expanding the known virome of mosquitoes in the Amazon. The identification of multiple novel viral species, alongside new records of viruses in the region, underscores the immense, yet largely unexplored, viral diversity circulating in these ecosystems. These findings highlight the critical importance of large-scale surveillance studies in biodiverse and ecologically dynamic areas, not only to advance our understanding of virus-mosquito interactions but also to provide essential insights into viral evolution and potential emerging viruses that could be important for human and animal health.
Supplemental Information
10.7717/peerj.20880/supp-1Supplemental Information 1Sampling sites where mosquito pools were collectedCoordinates for each sampling site are shown, as well as year of collection, number of pools, and total number of individuals
10.7717/peerj.20880/supp-2Supplemental Information 2Summary of RNA viral genomes and RdRp contigs recovered from mosquito poolsViral metagenome-assembled genomes (vMAGs) and RdRp-containing contigs identified in this study. For each sequence, we report the provisional virus name, best BLASTX hit (virus and protein accession), amino-acid identity (%), viral family, mosquito pool ID, strain ID, isolation year, GenBank accession, contig length (bp), segment (where applicable), expected set of genes, genes observed on the contig, CheckV completeness estimate (%), and the final completeness category (e.g. complete CDS, partial RdRp CDS, complete CDS for all three segments).An asterisk (* ) after the virus name denotes tentatively novel viruses described in this study. A double asterisk (**) indicates complete genomes recovered.
10.7717/peerj.20880/supp-3Supplemental Information 3Sequencing result details for each poolNumber of paired-reads before and after quality check are indicated, along with file names for the fastq files and BioSample IDs.
10.7717/peerj.20880/supp-4Supplemental Information 4Sequence clustering of Coredo-like virus sequences identified in this studyClustering was performed using CD-HIT with a 98% amino acid identity threshold. For each cluster, the representative sequence used for phylogenetic analysis is indicated, along with the sequences comprising each cluster and their amino acid identity to the representative.
10.7717/peerj.20880/supp-5Supplemental Information 5Sequence clustering of Jurua Merida virus sequences identified in this studyClustering was performed using CD-HIT with a 98% amino acid identity threshold. The representative sequence used for phylogenetic analysis is indicated, along with the sequences comprising the cluster and their amino acid identity to the representative.
10.7717/peerj.20880/supp-6Supplemental Information 6Amino acid identity matrix based on the RdRp protein sequence of Merida-like viruses identified in this study and reference sequencesColor shading represents the degree of identity, with darker blue indicating higher similarity.
10.7717/peerj.20880/supp-7Supplemental Information 7Amino acid identity matrix based on the RdRp protein sequence of strains from the Madalivirus genusColor shading represents the degree of identity, with darker blue indicating higher similarity.
10.7717/peerj.20880/supp-8Supplemental Information 8Amino acid identity matrix based on the RdRp protein sequence of the Culex flavivirus strains employed in this studyColor shading represents the degree of identity, with darker blue indicating higher similarity.
10.7717/peerj.20880/supp-9Supplemental Information 9Amino acid identity matrix based on the ORF1b protein sequence of the Mesonivirus strains employed in this studyColor shading represents the degree of identity, with darker blue indicating higher similarity.
10.7717/peerj.20880/supp-10Supplemental Information 10Amino acid identity matrix based on the RdRp protein sequence of the Totivirus strains employed in this studyColor shading represents the degree of identity, with darker blue indicating higher similarity.
10.7717/peerj.20880/supp-11Supplemental Information 11Viral genomes obtained in this studyViral genomes (complete and partial) and their respective annotations as they were submitted to the GenBank. Accession numbers assigned to the sequences can be found in Table 1. Most of these sequences are not publicly available yet
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Andrews S 2010 Babraham bioinformatics—Fast QC a quality control tool for high throughput sequence datahttps://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 2Auguste AJ Carrington CVF Forrester NL Popov VL Guzman H Widen SG Wood TG Weaver SC Tesh RB 2014 Characterization of a novel Negevirus and a novel Bunyavirus isolated from Culex (Culex) declarator mosquitoes in Trinidad The Journal of General Virology 95Pt 248148510.1099/vir.0.058412-024262627 PMC 3917067 · doi ↗ · pubmed ↗
- 3Azar SR Campos RK Bergren NA Camargos VN Rossi SL 2020 Epidemic alphaviruses: ecology, emergence and outbreaks Microorganisms 88116710.3390/microorganisms 808116732752150 PMC 7464724 · doi ↗ · pubmed ↗
- 4Barreto FK de A Alencar CH Araújo FM de C Oliveira R de MAB Cavalcante JW Lemos DRQ Farias LABG Boriz ILF Medeiros LQ Melo MNP Miyajima F Siqueira AM Freitas ARR Cavalcanti LP de G 2020 Seroprevalence, spatial dispersion and factors associated with flavivirus and chikungunya infection in a risk area: a population-based seroprevalence study in Brazil BMC Infectious Diseases 20188110.1186/s 12879-020-05611-533234110 PMC 7685300 · doi ↗ · pubmed ↗
- 5Bittar C Machado DC Vedovello D Ullmann LS Rahal P Araújo Junior JP Nogueira ML 2016 Genome sequencing and genetic characterization of Culex Flavirirus (Cx FV) provides new information about its genotypes Virology Journal 13115810.1186/s 12985-016-0614-327658621 PMC 5034531 · doi ↗ · pubmed ↗
- 6Bolling BG Weaver SC Tesh RB Vasilakis N 2015 Insect-specific virus discovery: significance for the arbovirus community Viruses 79910.3390/v 7092851 PMC 458429526378568 · doi ↗ · pubmed ↗
- 7Bray NL Pimentel H Melsted P Pachter L 2016 Near-optimal probabilistic RNA-seq quantification Nature Biotechnology 34552552710.1038/nbt.351927043002 · doi ↗ · pubmed ↗
- 8Buchfink B Reuter K Drost H-G 2021 Sensitive protein alignments at tree-of-life scale using DIAMOND Nature Methods 18436636810.1038/s 41592-021-01101-x 33828273 PMC 8026399 · doi ↗ · pubmed ↗