ACTA1‐Related Adult‐Onset Scapuloperoneal Myopathy With Cores and Rods
Alexandru Caramizaru, Marion Onnée, Sergey Nikitin, Amelia Dobrescu, Gianmarco Severa, Aysylu Murtazina, Andoni Urtizberea, Jean‐Pascal Lefaucheur, Robert‐Yves Carlier, Corinne Metay, Edoardo Malfatti

TL;DR
A new case of adult-onset muscle disease linked to a rare ACTA1 gene variant is described, showing a unique pattern of muscle weakness and structural changes.
Contribution
A novel ACTA1 variant (Pro334Leu) is identified in a late-onset scapuloperoneal myopathy with cores and rods, expanding the known clinical spectrum of actinopathies.
Findings
The patient had a slowly progressive muscle weakness pattern affecting proximal and distal limb muscles.
Muscle biopsy revealed cores and rods, and increased cardiac alpha-actin expression was observed.
The Pro334Leu ACTA1 variant is a previously unpublished pathogenic mutation associated with a milder actinopathy phenotype.
Abstract
Actinopathies are myopathies associated with pathogenic variants in ACTA1, a gene encoding the skeletal alpha‐actin protein. Although patients most frequently have a severe congenital myopathy, an important clinical and myopathological variability has been described. Recently, a scapuloperoneal myopathy phenotype associated with ACTA1 has been reported. Here, we present a Russian woman with a late‐onset, slowly progressive, scapuloperoneal actinopathy associated with an unpublished heterozygous pathogenic ACTA1 variant. We performed a thorough analysis of clinical, muscle imaging, muscle biopsy, genetic, protein and cardiac alpha‐actin expression data from a 65‐year‐old woman with a scapuloperoneal myopathy phenotype. Disease onset was at around 30 years with proximal lower limbs muscle weakness, which slowly progressed towards an upper and lower limb distal involvement with prominent…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
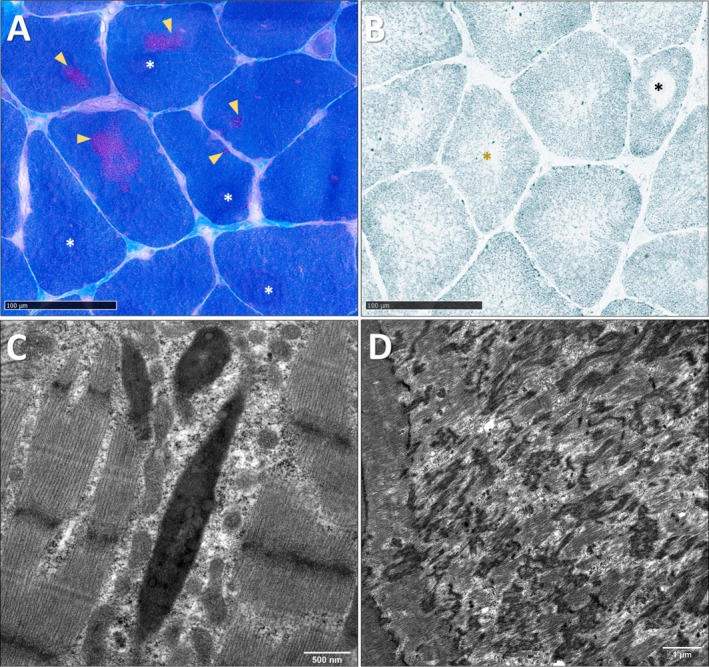 Figure 1
Figure 1 Figure 2
Figure 2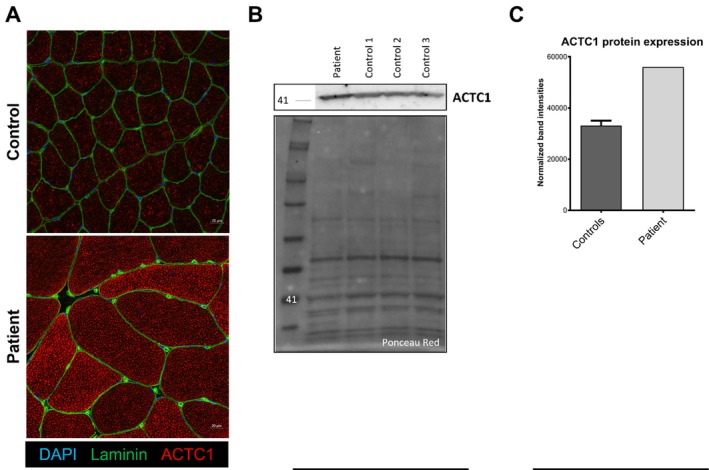 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Heterotopic Ossification and Related Conditions · Nuclear Structure and Function
The ACTA1 gene (1q42.13) encodes skeletal muscle alpha‐actin, a key contractile protein belonging to the actin family. Skeletal and cardiac alpha‐actin are the two sarcomeric isoforms expressed in muscle [1]. During foetal development, cardiac alpha‐actin predominates, but skeletal alpha‐actin becomes the main isoform by birth, while the postnatal heart primarily expresses cardiac alpha‐actin [2]. Cardiac involvement in actinopathies is rare [3, 4, 5], and even severe lethal cases usually allow survival to term [2]. Increased cardiac alpha‐actin expression in some long‐term survivors [6] suggests a compensatory role, mitigating the effects of ACTA1 variants [7].
Actinopathies are typically autosomal dominant [8] and clinically heterogeneous, ranging from severe congenital to milder adult‐onset forms [9]. Muscle biopsies show overlapping lesions, including actin accumulations, rods (intranuclear in 6% of cases), fibre‐type disproportion, cores, caps, dystrophic features, zebra bodies and lobulated fibres [3, 10]. Nemaline bodies (rods), derived from the Z‐line, are the hallmark of nemaline myopathy (NM) [11]. NM is most often caused by variants in NEB, encoding nebulin, a thin‐filament associated protein [12, 13, 14], and ACTA1, accounting for 50% and 20% of all NMs, respectively [15, 16].
ACTA1 has recently been linked to a novel scapuloperoneal myopathy phenotype, observed in 14 individuals within a single family [17]. This presentation features early scapuloperoneal and distal weakness, followed by mild facial involvement, Achilles tendon contractures and diminished reflexes, broadening the clinical spectrum of actinopathies.
Here, we report a patient with an adult‐onset slowly progressive scapuloperoneal myopathy related to a novel heterozygous pathogenic ACTA1 variant, NM_001100.4:c.1001C > T, p.(Pro334Leu), thus enlarging the landscape of ACTA1‐related disorders.
A 65‐year‐old woman, born to nonconsanguineous Russian parents, with no relevant family or early medical history, developed motor symptoms at 30, beginning with running difficulties and a stepping gait. By 48, she experienced progressive weakness affecting stair climbing, rising from a chair and bilateral finger extension (fourth–fifth digits). At 50, frequent falls occurred due to thigh weakness, especially when descending stairs.
Neurologic examination at 50 revealed right fourth finger extensor and quadriceps weakness (MRC 4), axial and mild distal leg weakness, thigh and gluteal atrophy, and reduced or absent reflexes, without sensory deficits. Serum CK was 360 U/L.
At 63, she showed a right‐predominant stepping gait, bilateral asymmetric scapula alata and limited lateral gaze, without other cranial involvement. Muscle testing demonstrated neck flexor (MRC 4) and proximal deltoid (MRC 4) weakness, severe extensor weakness for the fourth and fifth fingers bilaterally (MRC 2, Figure S1), and symmetric proximal–distal lower limb weakness involving the psoas, quadriceps and tibialis anterior (MRC 4).
A whole‐body muscle MRI, performed at the age of 64 (Figure S2), highlighted a predominant involvement of the tongue, quadriceps, soleus and tibialis anterior, with relative sparing of the gracilis and select hamstring muscles.
A deltoid muscle biopsy at 63 years of age, showed the presence of clusters of cytoplasmic rods indicated by yellow arrowheads, and cores in separate areas of the myofibres, indicated by white asterisks (Figure 1A). Oxidative histoenzymatic reaction NADH‐TR revealed the presence of both intermyofibrillar disorganisation (yellow asterisk) and well‐delimited cores (black asterisk, Figure 1B). There was a type 2 fibre predominance. Transmission electron microscopy ultrastructural studies confirmed the presence of Z‐line thickening forming typical rods (Figure 1C), characterised by orthogonal net disposition of filaments and the presence of well‐defined areas of disorganisation and Z‐line material accumulation, corresponding to typical, unstructured cores; there are no mitochondria visible in the core area (Figure 1D).
A heterozygous missense variant was identified in exon 7 of ACTA1 (NM_001100.4:c.1001C > T, p.(Pro334Leu)), characterised as Class 4, Likely Pathogenic according to the 2015 ACMG guidelines [18].
We performed cardiac muscle alpha‐actin (ACTC1) protein expression analysis (Figure 2) by immunostaining using anti‐ACTC1 antibody, highlighting an overexpression of cardiac alpha‐actin in the patient's biopsy compared to controls. This observation was confirmed by western blot analysis. We could also observe a significant fibre hypertrophy in the patient's muscle biopsy compared to the control.
Detailed results (clinical, MRI, biopsy, genetic, protein and cardiac alpha‐actin expression) are available in Table S1.
Actinopathies usually manifest as severe congenital myopathies with hypotonia, weakness, facial involvement and respiratory or feeding difficulties, though adult‐onset forms also occur [3]. Nemaline myopathies show similar variability [11], with ACTA1 variants accounting for over half of severe cases [6]. Our patient's onset at 30 years is therefore atypical.
In the previously reported family with ACTA1‐related scapuloperoneal myopathy (c.591G > T, p.(Glu197Asp)) [17], disease onset and severity varied, with distal‐to‐proximal leg weakness and early scapular winging, deltoid/thenar atrophy, and distal finger extensor deficits,partially resembling our patient's finger extensor weakness. However, our patient showed initial proximal lower limb involvement followed by distal progression.
Other ACTA1‐related distal myopathies have similarly featured finger flexor and extensor weakness, particularly in digits 3–5, though onset and course differ among families. Therefore, such findings should be interpreted within the broader clinical and familial context when guiding diagnosis [19, 20].
MRI studies of ACTA1‐related myopathies show characteristic thigh involvement of the vasti, sartorius and biceps femoris, with relative sparing of the adductors and gracilis. In the lower legs, the soleus and tibialis anterior are typically affected, while the gastrocnemii and tibialis posterior remain relatively preserved [21, 22]. In the family with ACTA1‐related scapuloperoneal myopathy, one patient showed marked atrophy and significant fatty replacement of the quadriceps, while severe involvement was noted in the hamstring muscles, with partial sparing of semitendinosus and adductor longus muscles; a milder case showed minimal changes in the sartorius, biceps femoris and vastus lateralis [17]. Notably, fibrofatty tongue replacement may also occur in actinopathies without dysphagia [23], a rare finding outside disorders such as Duchenne muscular dystrophy, facioscapulohumeral dystrophy [24], oculopharyngeal muscular dystrophy [25] or late‐onset Pompe disease [26]. Our patient's MRI broadly reflected these patterns, highlighting notable interindividual variability.
ACTA1 variants cause a spectrum of myopathies (including intranuclear rod, actin‐accumulation, cap myopathy and congenital fibre‐type disproportion) [27, 28, 29, 30], characterised by overlapping histological features such as actin aggregates, rods, caps and core‐like areas [3]. Severe neonatal cases often show type I fibre atrophy with rods, cores or cytoplasmic bodies [6]. Our patient's biopsy revealed rods and cytoplasmic cores, consistent with this morphopathologic variability, and also partly resembling core‐rod myopathy [31]; however, the clinical phenotype is not supportive of this diagnosis, although ACTA1 variants have been linked to core and rod pathology [32, 33, 34].
Muscle pathology in ACTA1‐related scapuloperoneal myopathy ranges from marked fibre size variability, nuclear internalisation and fatty infiltration to mild changes without rods or cores. Notably, nemaline rods, cores or actin aggregates were absent in these cases [17], contrasting with our patient's biopsy.
A significant variability has also been reported in distal myopathies with finger weakness, which may show, among other changes, rods, cores or fibre‐type disproportion [19, 20, 35]. These findings are overall similar to the ones described in our patient.
Around 90% of ACTA1 variants are autosomal dominant missense changes, while recessive cases typically result from null mutations [3]. The NM_001100.4:c.1001C > T (p.(Pro334Leu)) variant affects a conserved residue, is absent from population databases, and is predicted to be deleterious (REVEL 0.814; CADD 32) [36]. ACTA1 shows low tolerance for benign missense variation, with pathogenic missense changes representing a common disease mechanism (ClinGen Congenital Myopathies VCEP ‐ Variant Curation Expert Panel). This variant has been previously reported in ClinVar, albeit with limited clinical context. Notably, a similar substitution at this codon (p.(Pro334Arg)) has been reported as likely pathogenic in a late‐onset dominant alpha‐actinopathy with consistent muscle biopsy findings (Invitae, Revvity Omics, CeGaT and Paris‐East Créteil University internal data, Alan Beggs personal communication, Leiden Open Variation Database). Accordingly, the evidence supports the classification of p.(Pro334Leu) as likely pathogenic under current ACMG/AMP criteria.
Variants near Pro334 (e.g., p.(Pro332Ser)) cause severe congenital fibre‐type disproportion with marked weakness and respiratory involvement but preserved ocular motility [37]; our patient's mild left‐gaze limitation therefore diverges from this pattern.
Since severe early‐onset phenotypes often correlate with variants in exons 2–5 [6], the milder, late‐onset presentation in our patient may reflect the variant's location in exon 7 [1]; however, reports of mild phenotypes with upstream variants [19, 20, 35] suggest that additional modifying factors likely contribute.
In survivors of severe ACTA1‐related nemaline myopathy beyond the neonatal period [6], increased cardiac alpha‐actin expression has been reported as a potential compensatory mechanism. Our patient similarly demonstrates cardiac alpha‐actin expression, possibly contributing to the milder phenotype. Although such upregulation is rarely described in thin‐filament myopathies outside ACTA1 variants [38, 39], persistence of developmental proteins (including cardiac alpha‐actin) has been observed in severe congenital myopathies caused by MYL1 and other thick‐filament gene defects, particularly with respect to myosin isoform expression [40]. These observations suggest that cardiac alpha‐actin expression may modulate disease severity in actinopathies.
With this patient, we expand the clinical and genetic spectrum of ACTA1 myopathies. ACTA1‐related congenital myopathies remain a clinically, genetically and histopathologically variable group of disorders. Particularly, the scapuloperoneal phenotype could represent a distinct subcategory, and the characterisation of this patient with a less severe, different clinical presentation and nemaline bodies should contribute to its understanding.
Author Contributions
E.M. designed the study. S.N., E.M., G.S. and M.O. performed the study and analysed the data. M.O. performed the experiments. S.N., E.M. and G.S. provided clinical data and provided resources. A.C., M.O., S.N. and G.S. wrote the manuscript. E.M. supervised the study. The manuscript was revised by E.M., C.M., R.C., J.L., A.U., A.D., A.M. and S.N.
Funding
The authors have nothing to report.
Ethics Statement
This study was approved by the ethical Committee issued by our institutions in compliance with the Declaration of Helsinki. Informed consent has been obtained from the patient for participation and muscle biopsy studies, according to the French Comité de Protection des Personnes Est IV DC‐2012‐1693.
Consent
Informed consent for publication, including pictures, has been obtained from the patient.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1: Weakness of the extensors of the fourth and fifth fingers of the hands.
Figure S2: Muscle imaging of multiple sequential T1W and STIR MRI acquisitions in the axial plan. CRANIAL sections (A, B): fibrofatty infiltration of tongue (A, arrow). AXIAL sections (C–F): focal left deltoid involvement at the medial side (C, arrow) and bilateral substitution of paraspinal muscle at the lumbar level (E, arrow). LOWER LIMBS sections (G–N): severe and bilateral fibrofatty substitution of quadriceps and biceps femoris long head (I, arrows) associated with quadriceps muscle hypersignal in STIR sequences (J, yellow asterisk). Gastrocnemius medialis and tibialis anterior involvement (K and M, arrows); Soleus hypersignal in STIR (N, yellow asterisk).
Figure S3: Two‐dimensional and three‐dimensional ACTA1 protein structure. (A) Localisation of ACTA1 c.1001C > T, p.(Pro334Leu) variant is indicated in red on the ACTA1 gene and protein subdomains. ‘SD’ indicates the different ACTA1 subdomains, which are colour‐coded to correspond to the colours seen throughout the 3D protein structure in Panel C. The hinge domains are represented in grey and correspond to amino acids 137 to 150 and 333 to 338. (B) The Pro334Leu missense variant is located in a highly conserved region among 11 different species. (C) ACTA1 is divided into two domains connected by two hinge domains; each ACTA1 domain is further divided into two subdomains (SD 1 and 2; and SD 3 and 4). Subdomains‐coloured model of ACTA1 monomer was built using AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/; entry P68133). The residue 334 is shown in magenta (Pro334 in the left panel and mutated Leu334 in the right panel). The residues at < 5 Å of residue 334 are shown in stick representation with coloured atoms (H in white, O in red, and N in blue). Hydrophobic interactions are shown by yellow lines.
Figure S4: Cryo‐EM structure of recombinant wild‐type ACTA1 phalloidin‐stabilised F‐actin (light pink) (PDBID 9DUU on https://www.rcsb.org/). One ACTA1 monomer is coloured in yellow, blue, orange and green, representing its different subdomains (SD1, SD2, SD3 and SD4, respectively), the hinge domain is represented in light grey. The residue Pro334 is represented in magenta. We can notice that the Pro334 residue is in the outside part of the F‐actin, likely interacting more with other proteins of the thin filament rather than with other ACTA1 monomers.
Table S1: Detailed clinical, muscle MRI, muscle biopsy, genetic, protein and cardiac alpha‐actin expression data regarding a patient with an ACTA1‐related adult‐onset scapuloperoneal myopathy with cores and rods.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1N. G. Laing , D. E. Dye , C. Wallgren‐Pettersson , et al., “Mutations and Polymorphisms of the Skeletal Muscle Alpha‐Actin Gene (ACTA 1),” Human Mutation 30, no. 9 (2009 Sep): 1267–1277.19562689 10.1002/humu.21059 PMC 2784950 · doi ↗ · pubmed ↗
- 2B. Ilkovski , S. Clement , C. Sewry , K. N. North , and S. T. Cooper , “Defining Alpha‐Skeletal and Alpha‐Cardiac Actin Expression in Human Heart and Skeletal Muscle Explains the Absence of Cardiac Involvement in ACTA 1 Nemaline Myopathy,” Neuromuscular Disorders 15, no. 12 (2005 Dec): 829–835.16288873 10.1016/j.nmd.2005.08.004 · doi ↗ · pubmed ↗
- 3K. J. Nowak , G. Ravenscroft , and N. G. Laing , “Skeletal Muscle α‐Actin Diseases (Actinopathies): Pathology and Mechanisms,” Acta Neuropathologica 125, no. 1 (2013 Jan): 19–32.22825594 10.1007/s 00401-012-1019-z · doi ↗ · pubmed ↗
- 4A. Garg , S. Jansen , R. Zhang , et al., “Dilated Cardiomyopathy‐Associated Skeletal Muscle Actin (ACTA 1) Mutation R 256H Disrupts Actin Structure and Function and Causes Cardiomyocyte Hypocontractility,” bio Rxiv. (2024) Mar 12.10.1073/pnas.2405020121 PMC 1157296939503885 · doi ↗ · pubmed ↗
- 5A. Matsumoto , H. Tsuda , S. Furui , et al., “A Case of Congenital fiber‐Type Disproportion Syndrome Presenting Dilated Cardiomyopathy With ACTA 1 Mutation,” Molecular Genetics & Genomic Medicine 10, no. 9 (2022 Sep): e 2008.35757965 10.1002/mgg 3.2008 PMC 9482392 · doi ↗ · pubmed ↗
- 6C. Labasse , G. Brochier , A. L. Taratuto , et al., “Severe ACTA 1‐Related Nemaline Myopathy: Intranuclear Rods, Cytoplasmic Bodies, and Enlarged Perinuclear Space as Characteristic Pathological Features on Muscle Biopsies,” Acta Neuropathologica Communications 10, no. 1 (2022): 101.35810298 10.1186/s 40478-022-01400-0PMC 9271256 · doi ↗ · pubmed ↗
- 7G. Ravenscroft , E. Mc Namara , L. M. Griffiths , et al., “Cardiac α‐Actin Over‐Expression Therapy in Dominant ACTA 1 Disease,” Human Molecular Genetics 22, no. 19 (2013): 3987–3997.23736297 10.1093/hmg/ddt 252 · doi ↗ · pubmed ↗
- 8M. Ogasawara and I. Nishino , “A Review of Major Causative Genes in Congenital Myopathies,” Journal of Human Genetics 68, no. 3 (2023 Mar): 215–225.35668205 10.1038/s 10038-022-01045-w · doi ↗ · pubmed ↗