Virtual Screening of Marine Natural Products Targeting the F Protein for Anti-RSV Drug Discovery
Wenqing Liu, Xuran Gu, Ruikun Du, Zhiqing Liu, Pingyuan Wang, Chang-Yun Wang

TL;DR
This study uses virtual screening to find marine natural products that could inhibit RSV by targeting its F protein, with manzamine alkaloids showing strong potential.
Contribution
The study introduces marine natural products, particularly manzamine alkaloids, as novel leads for RSV fusion inhibitors.
Findings
11 promising compounds were identified from 31,561 marine natural products via virtual screening and ADMET profiling.
Manzamine alkaloids showed the best docking scores and favorable interactions with conserved residues on the F protein.
The compounds interact through hydrophobic, π-stacking, and electrostatic interactions with the F protein.
Abstract
Respiratory syncytial virus (RSV) poses a substantial global health burden, particularly in infants and the elderly. The fusion (F) protein is a key therapeutic target for inhibiting RSV entry. In this study, we performed a structure-based virtual screening of the Comprehensive Marine Natural Products Database (CMNPD) to discover novel anti-RSV agents targeting the prefusion F protein trimer. Screening of 31,561 compounds via molecular docking, followed by stringent ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiling and MM/GBSA (Molecular Mechanics/Generalized Born Surface Area) binding free energy calculations, identified 11 promising candidates. Among these, manzamine alkaloids exhibited the most favorable docking scores (as low as −13.3 kcal/mol) and promising Ligand Efficiency (LE) values. These molecules primarily interact with conserved hydrophobic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
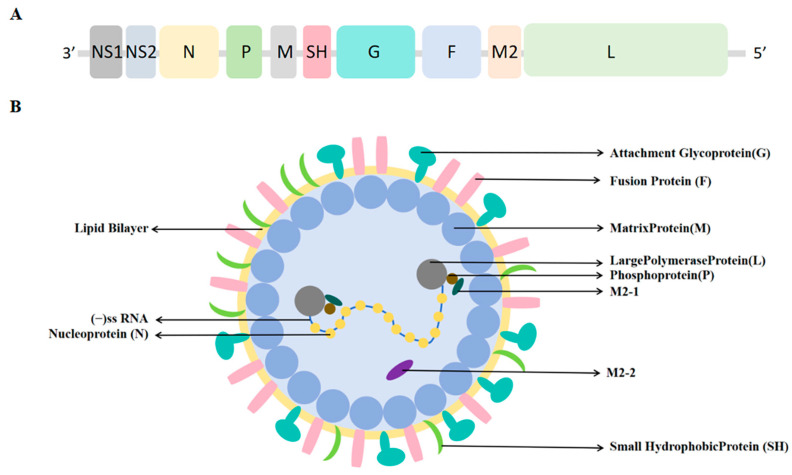 Figure 1
Figure 1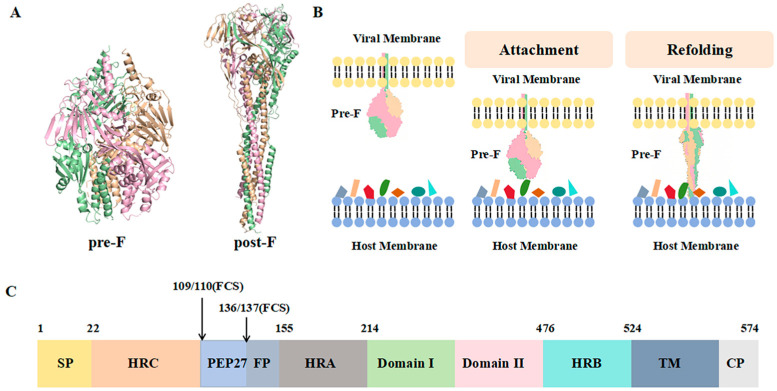 Figure 2
Figure 2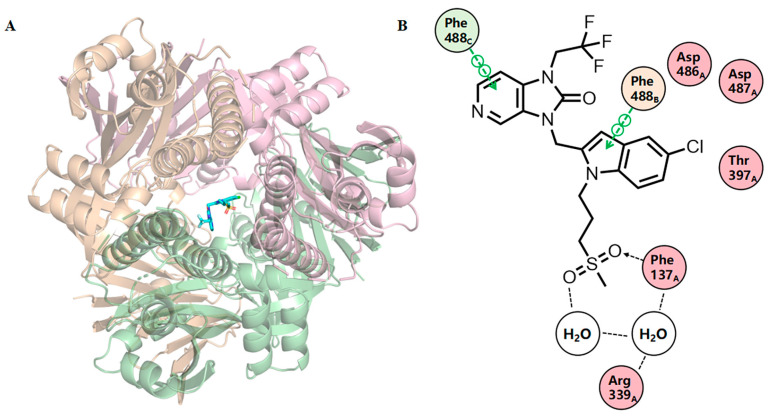 Figure 3
Figure 3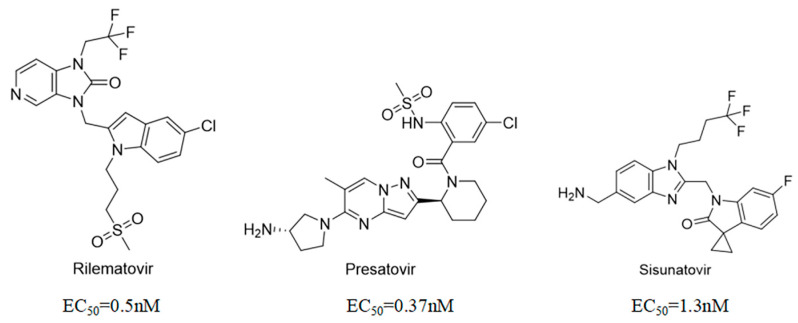 Figure 4
Figure 4 Figure 5
Figure 5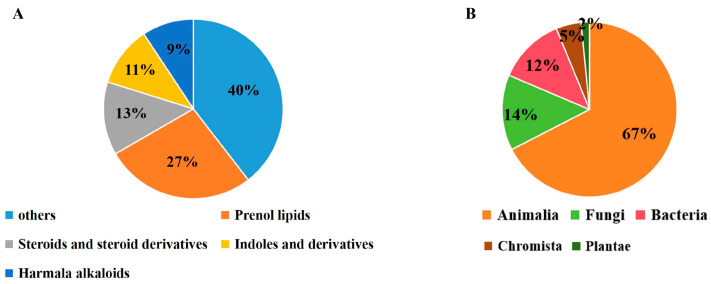 Figure 6
Figure 6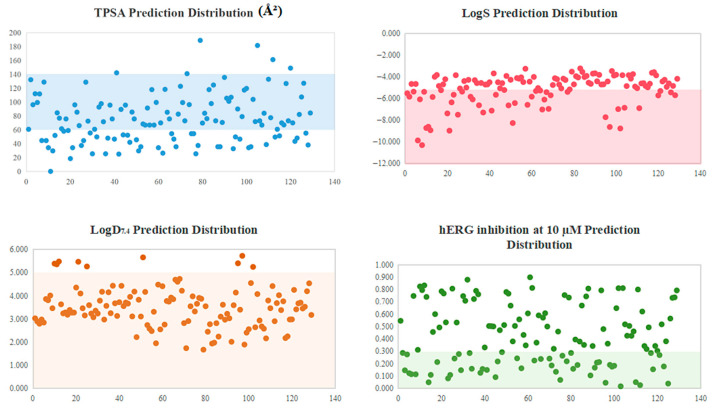 Figure 7
Figure 7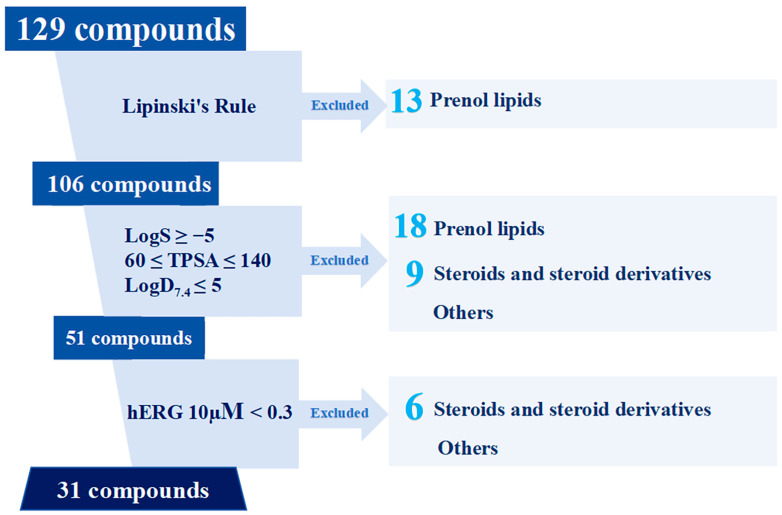 Figure 8
Figure 8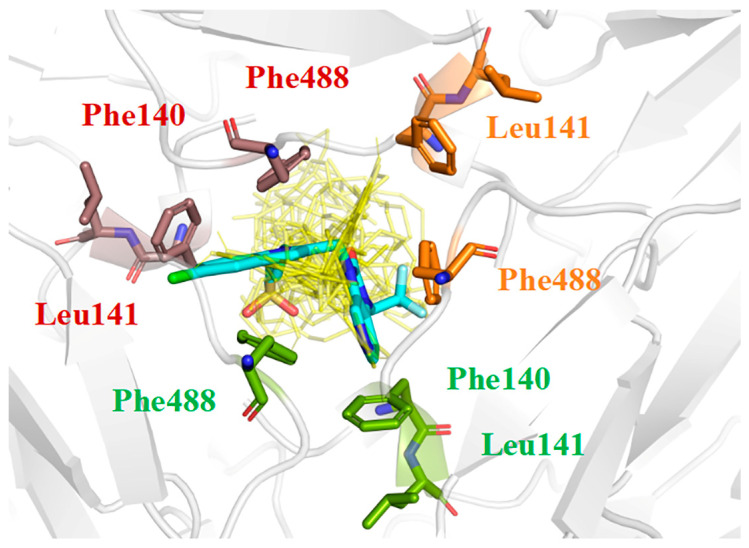 Figure 9
Figure 9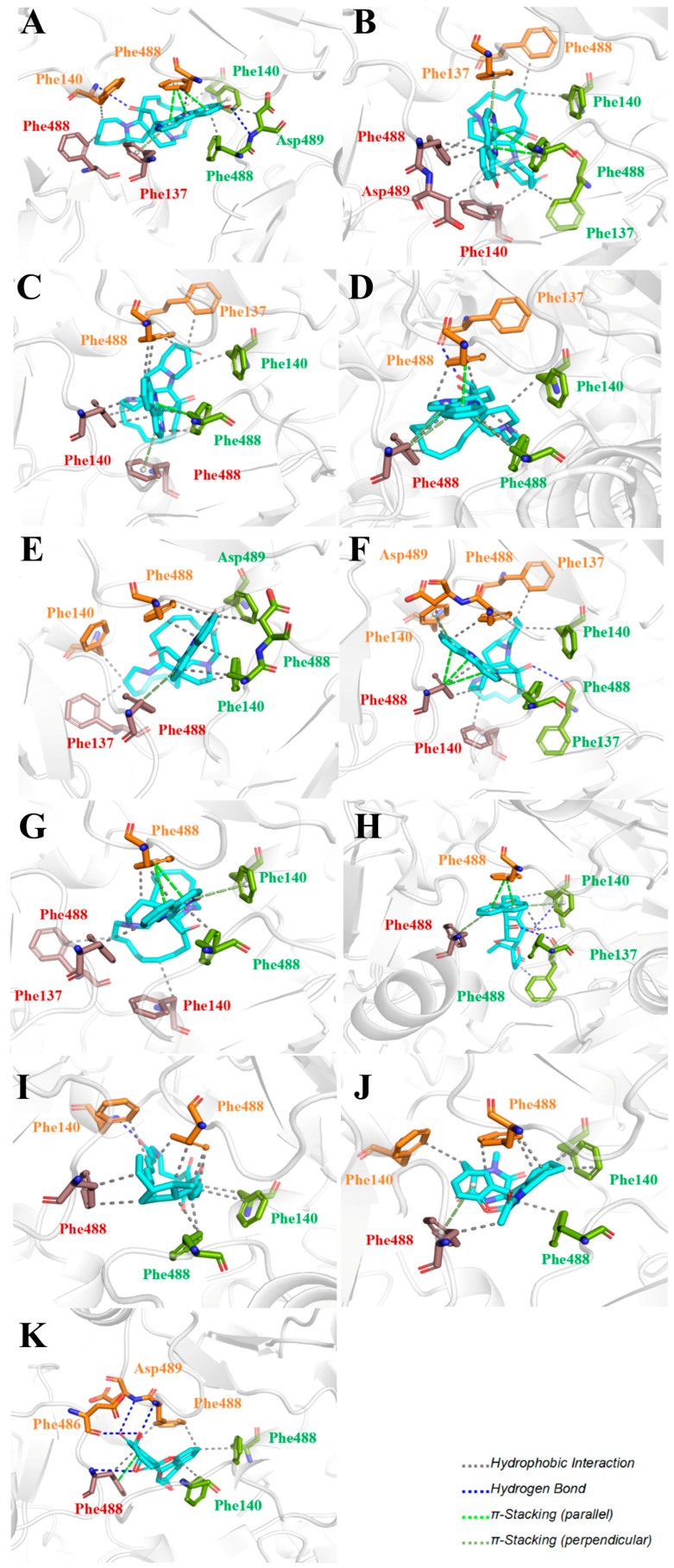 Figure 10
Figure 10 Figure 11
Figure 11 Figure 12
Figure 12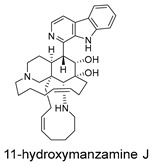 Figure 13
Figure 13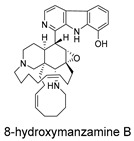 Figure 14
Figure 14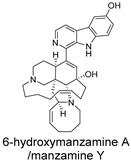 Figure 15
Figure 15 Figure 16
Figure 16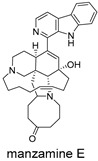 Figure 17
Figure 17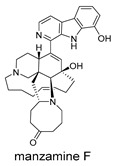 Figure 18
Figure 18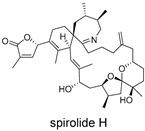 Figure 19
Figure 19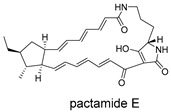 Figure 20
Figure 20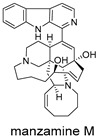 Figure 21
Figure 21 Figure 22
Figure 22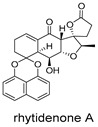 Figure 23
Figure 23 Figure 24
Figure 24 Figure 25
Figure 25 Figure 26
Figure 26 Figure 27
Figure 27 Figure 28
Figure 28 Figure 29
Figure 29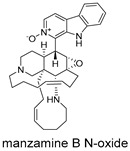 Figure 30
Figure 30 Figure 31
Figure 31 Figure 32
Figure 32 Figure 33
Figure 33 Figure 34
Figure 34 Figure 35
Figure 35 Figure 36
Figure 36 Figure 37
Figure 37 Figure 38
Figure 38 Figure 39
Figure 39 Figure 40
Figure 40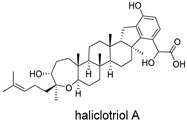 Figure 41
Figure 41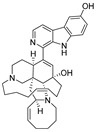 Figure 42
Figure 42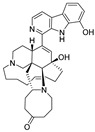 Figure 43
Figure 43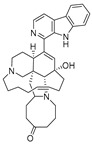 Figure 44
Figure 44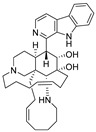 Figure 45
Figure 45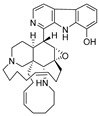 Figure 46
Figure 46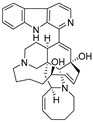 Figure 47
Figure 47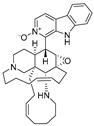 Figure 48
Figure 48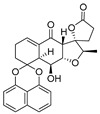 Figure 49
Figure 49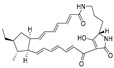 Figure 50
Figure 50- —National Natural Science Foundation of China
- —Natural Science Foundation of Shandong Province
- —Taishan Scholars Program of Shandong Province, China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRespiratory viral infections research · vaccines and immunoinformatics approaches · SARS-CoV-2 and COVID-19 Research
1. Introduction
Respiratory syncytial virus (RSV) is a highly seasonal pathogen that primarily infects the respiratory tract and exhibits strong pathogenicity, frequently causing severe illnesses such as pneumonia and bronchiolitis [1]. While most infections are mild and self-limiting, the virus can be severe in young children, immunocompromised adults, and the elderly, potentially progressing to fatal respiratory failure [2,3]. Given its profound impact, RSV is recognized as a major public health concern, necessitating effective preventive and therapeutic strategies [4].
Currently approved clinical therapeutics for RSV primarily consist of prophylactic drugs such as monoclonal antibodies, including monoclonal antibody therapies such as Nirsevimab (Beyfortus) and Clesrovimab (Enflonsia) [5,6], and RSV vaccines like GSK’s Arexvy, Pfizer’s Abrysvo and Moderna’s mRNA vaccine mRESVIA (mRNA-1345) [7]. Their utility is primarily prophylactic, preventing infection initiation, but often ineffective against established infections. Hence, the development of small-molecular drugs remains among the key priorities for anti-RSV agent development [8].
RSV is a non-segmented, negative-sense, single-stranded, and enveloped RNA virus [9]. Its genome encodes 11 proteins, including the attachment glycoprotein (G), fusion protein (F), and small hydrophobic protein (SH) in the envelope, as well as internal structural (N, P, L, M2-1, M2-2) and non-structural (NS1, NS2) proteins (Figure 1) [10]. Viral entry is mediated by G protein attachment and F protein-driven, clathrin-dependent endocytosis [11].
The F protein (Figure 2) represents one of the most promising therapeutic targets for anti-RSV drug design. Significant progress has been made in recent years toward elucidating the binding mode and inhibitory mechanism of fusion inhibitors targeting the F protein [12]. F protein inhibitors block viral infection by preventing the fusion of the viral envelope with the host cell membrane. The F protein exists in two distinct conformational states: the prefusion conformation (pre-F) and the postfusion conformation (post-F) (Figure 2A) [13]. The pre-F conformation represents a high-energy, metastable state that readily undergoes structural rearrangement to transition into the more stable, low-energy post-F conformation (Figure 2B) [14]. This critical step is mediated by the six-helix bundle domain of the F protein, a fusion mechanism characteristic of the Paramyxoviridae family. To date, most RSV F inhibitors developed for therapeutic purposes target the pocket deeply embedded within the central cavity of the pre-F trimer [15], thereby preventing transition to the postfusion state and the formation of its characteristic six-helix bundle. Before virus entry, the F protein trimer is maintained in the prefusion conformation, with the fusion peptide deeply buried in the protein interior and inaccessible to solvents. After a series of conformational changes, the F protein’s fusion peptide is exposed and inserted into the target cell membrane. Subsequently, the HRB and HRA domains interact to form a stable six-helix bundle (6-HB) core structure, resulting in membrane apposition. Finally, the fusion pore opens, allowing for the entry of the viral genetic material into the target cell [16].
The most advanced F protein inhibitor, Rilematovir (JNJ-53718678), exhibits direct binding to pre-F [17]. It progressed to phase 3 clinical trials following the demonstration of potent in vitro activity (EC_50_ = 0.5 nM against recombinant rgRSV224 in HeLa cells). However, its clinical development has been terminated. The co-crystal structure revealed that Rilematovir asymmetrically occupies the binding pocket (Figure 3A) and engages in aromatic π-stacking interactions with Phe140 and Phe488 residues of the F protein. In the binding site (Figure 3B), the two fused rings engage in extensive π-π stacking and weak C-H/π interactions with aromatic residues such as Phe140 and Phe488, forming a hydrophobic binding core. The establishment of these aromatic protein–ligand stacking interactions appears to be a conserved feature among all known RSV fusion inhibitors and likely restricts the central heterocyclic moieties of these inhibitors to a fixed conformation [15].
Well-defined protein structures, clearly characterized binding sites, and elucidated mechanisms of action have collectively accelerated rapid progress in related research. Multiple RSV F inhibitors have now advanced into the clinical pipeline, for example, Rilematovir, Sismatovir (RV521) (mean IC_50_ = 1.3 nM vs. RSV-A/B strains in Balb/c mice) [18], and Presatovir (GS-5806) (mean EC_50_ = 0.37 nM, range 0.15–1.09 nM, vs. rgRSV-A/HEp-2) [19] (Figure 4). However, currently reported small-molecule inhibitors targeting the F protein exhibit limited structural diversity. Multiple reports have been published to elucidate the drug resistance of this class of compounds [20,21]. Research has revealed that both K394R and K394H mutations in the F protein confer cross-resistance to RSV fusion inhibitors. For example, the single K394R mutation in the F protein confers 1250-fold resistance to BMS-433771, while it confers 6024-fold increased resistance to Rilematovir [22].
Marine natural products (MNPs) represent a rich source of structurally novel bioactive compounds [23]. Marine natural products have proven to be effective biological modulators, with several marine-derived compounds already approved for clinical use [24]. Their unique structures and bioactivities continue to drive interest in their application for drug discovery.
Computer-aided drug design (CADD) is an emerging field that has drawn considerable interest because of its potential to expedite and lower the cost of the drug development process [25]. Drug discovery is an expensive and time-consuming process, frequently taking 10–15 years for a drug to reach the market. Given these challenges, CADD has substantially changed research in the field by reducing the time and cost associated with drug development [26].
In this study, we applied a CADD strategy to systematically screen the Comprehensive Marine Natural Products Database (CMNPD, https://cmnpd.org/) [27] for novel F-protein inhibitors [28]. The process involved molecular docking, ADMET filtering, and MM/GBSA binding free energy calculations to identify promising, drug-like candidates. Through integration of all results, compounds exhibiting potential anti-RSV activity and optimal drug-like properties were identified.
2. Results
All 31,561 molecules in the CMNPD were initially screened for their target binding poses and scores using molecular docking (Figure 5). The top ranked compounds were subsequently screened using integrated absorption, distribution, metabolism, excretion, and toxicity (ADMET) predictions [29]. Considering the inherent limitations of molecular docking, which operates on highly simplified computational models and performs basic calculations primarily based on shape complementarity between ligands and the protein binding pocket, only a narrow range of interactions were considered in the scoring function. Consequently, MM/GBSA was employed in conjunction with molecular docking to refine the estimation of binding energies. A significant strength of this approach lies in its capacity to treat both the ligand and the protein with flexibility, thereby accommodating the structural adaptations necessary for induced fit binding. MM/GBSA additionally incorporated more comprehensive energy terms, such as gas-phase molecular mechanical energy, solvation free energy, and entropy changes. The calculated total MM/GBSA binding free energy was decomposed into per-residue contributions. MM/GBSA calculations were additionally performed to enable a more in-depth analysis of their binding modes [30,31].
2.1. Molecular Docking
Molecular docking was conducted using AutoDock Vina to evaluate the anti-RSV potential of MNPs targeting the F protein (chains A, B, C). The co-crystal structure of the F protein with Rilematovir (PDB code: 5KWW) is depicted in Figure 3 [15]. All 31,561 MNPs (47,450 configurations) in the CMNPD were docked. The conformation with the lowest binding energy for each ligand was regarded as the most favorable conformation. The results for compounds with binding energies below −12.0 kcal/mol are compiled in Table S1. The co-crystallized Rilematovir and Sisunatovir, two known RSV F inhibitors, were used as positive controls.
A total of 129 compounds exhibited binding energy ranging from −15.9 to −12 kcal/mol, all lower than those of Rilematovir (−8.2 kcal/mol) and Sisunatovir (−10.5 kcal/mol). Chemically, the compounds primarily belong to indoles and derivatives (14), harmala alkaloids (12), prenol lipids (35), and steroids and steroid derivatives (17) (Figure 6A), sharing an extended rigid hydrophobic core strategically incorporated with heteroatoms. These compounds were derived from diverse biological kingdoms. The majority were from Animalia (87), followed by Fungi (18), Bacteria (16), Chromista (6), and Plantae (2) (Figure 6B). Most compounds were obtained from marine invertebrates, specifically sponges and corals. Fungal-origin compounds were isolated from Aspergillus, Penicillium, and Guignardia genera. Bacterial compounds were nearly all from Streptomyces species, classic producers of antibiotics and halogenated compounds.
2.2. Lipinski’s Rule of Five and ADMET Profile
The 129 compounds were evaluated for drug-likeness using Lipinski’s rules and ADMET prediction. Their properties, including rule compliance and ADMET predictions are summarized in Table S2.
Preliminary filtering applied Lipinski’s Rules: molecular weight ≤ 500 Da; calculated octanol–water partition coefficient (LogP) ≤ 5; hydrogen bond acceptors ≤ 10; hydrogen bond donors ≤ 5; estimated oral absorption ≥ 30% [29]. The final criterion was not strictly applied due to the inherent challenges in accurately predicting oral absorption in silico. It was found that 18 out of the 129 compounds violated more than two rules and were thereby excluded from further consideration.
ADMET properties were predicted using ADMETlab 3.0 [32]. Drug-likeness is largely determined by key properties such as aqueous solubility, membrane permeability, and safety profiles. To capture these critical aspects, four parameters—logS, LogD_7.4_, TPSA, and hERG inhibition at 10 µM—were selected for compound screening (Figure 7). Compounds with logS < −5 have poor water solubility [33]. LogD_7.4_ builds on logP by incorporating physiological pH 7.4 [34]; TPSA 60–140Å^2^ correlates with adequate membrane permeability and oral absorption. hERG inhibition risk was assessed to prevent potential cardiotoxicity (drug-induced torsades de pointes) [35]. Integrating these parameters, compounds with superior drug-like properties were prioritized [36]. Of the 129 candidates, 106 (82.2%) complied with Lipinski’s Rule of Five (RO5). Among these, 71 (67.0%) met the LogS ≥ −5; 51 (71.8% of solubility-qualified) had appropriate TPSA (60–140Å^2^); and 31 (60.8% of TPSA-qualified) showed low hERG risk (inhibition < 30% at 10 µM). Finally, 31 compounds complying with Lipinski’s Rule and meeting the criteria (logS ≥ −5, logD_7.4_ < 5, TPSA 60–140 Å^2^, hERG inhibition < 0.3 at 10 µM) were selected (Table 1). The predicted ADMET properties for all 129 compounds are visualized in Figure 7.
Based on ADMET predictions, 31 drug-like compounds were selected from the 129 compounds (Figure 8). Among these, indoles and derivatives (8) and harmala alkaloids (7) collectively accounted for half, remaining the most prevalent classes. The chemical classes of compounds excluded during the screening process based on Lipinski’s rule and ADMET criteria are shown in Figure 8.
Compound classes such as azepines, macrolactams, and naphthofurans exhibited minimal change in numbers after selection, indicating promising ADMET profiles. In contrast, only 1 of the 35 prenol lipids remained after screening, and only 2 of the 17 steroids remained. Detailed analysis revealed that the elimination of prenol lipids was primarily due to extremely poor aqueous solubility (high molecular weight and lipophilicity). A total of 13 molecules violated Lipinski’s Rule; of the remaining 22, 18 were excluded due to unfavorable TPSA and logS, and of the final 4, 3 were filtered out due to high hERG risk. Only CMNPD11217 met all criteria (Table 1). For steroids, high cardiotoxicity potential was the major cause of elimination; only 5 out of 17 had hERG < 0.3, while 3 of these 5 exhibited high lipophilicity, resulting in only 2 ultimately meeting all criteria.
The molecular weights of the 31 compounds range from 360 to 580 g/mol, with some exceeding the 500 g/mol benchmark for oral drugs. Since the positive control drug Rilematovir also exceeds this benchmark, we preliminarily considered these compounds within the common drug-like molecular weight range. Predicted logS values (−3.2 to −4.8) fall into the “soluble” to “moderately soluble” categories [37]. LogD_7.4_ values (1.7 to 3.7) indicate an optimal balance between hydrophilicity and lipophilicity, supporting both aqueous solubility and membrane permeability [38]. All TPSA values were below the 140 Å^2^ permeability threshold, with most under 100 Å^2^, suggesting good membrane permeability potential [39]. Predicted hERG inhibition probabilities are all <0.3, with over 60% (19 molecules) below 0.2, suggesting a favorable cardiac safety profile that mitigates cardiotoxicity risk in later development [36]. These compounds exhibit drug-likeness comparable to the positive controls.
2.3. Interaction Analysis
A detailed analysis of interactions between the 31 compounds and the F protein was conducted, identifying specific amino acid residues and interaction types (Table 2).
All 31 compounds interact with a critical set of conserved residues, with hydrophobic interactions being the dominant binding force. Hydrophobic interactions were observed for all compounds, primarily with Phe488, Phe140, and Phe137. Hydrogen bonding occurred in 22 compounds (~70%) involving residues such as Phe140, Gly139, Asp486, and Gln354. π-Stacking interactions were observed in 22 molecules, primarily with Phe140 and Phe488, stabilizing aromatic systems within the binding pocket. Notably, CMNPD31296 forms a salt bridge with Arg339C, indicating higher binding specificity and potential. Fourteen compounds engaged in all three interaction types (hydrophobic, hydrogen bonding, π-stacking) which may contribute to higher binding affinity and effective stabilization of the binding pocket. CMNPD25781 binds solely via hydrophobic interactions, underscoring the potency of hydrophobic interactions for this target.
2.4. MM/GBSA Binding Free Energy Calculation and Integrated Compound Assessment
The MM/GBSA approach was employed to calculate the binding free energy for the 31 molecules, serving as a rescoring parameter to optimize compounds identified from docking [31]. The results are summarized in Table 3, and the complete dataset is available in Table S3 in the Supplementary Materials.
The binding free energy (ΔGbind) was decomposed into molecular mechanics energy components to elucidate individual contributions. More negative values indicate stronger attractive forces; positive values represent unfavorable effects. Ligand Efficiency (LE) was also evaluated [40]. It is generally accepted that a ligand with |LE| ≥ 0.3 kcal·mol^−1^ per non-H atom indicates high efficiency and improved drug-likeness. All 31 compounds met this criterion, with |LE| ranging from 0.87 to 2.65, demonstrating effective target binding with minimalist structures. Total ΔGbind ranged from −111.15 to −34.04 kcal/mol, significantly different from the docking energy range (−15.9 to −12 kcal/mol), indicating that MM/GBSA provides a more reliable binding assessment.
Based on primary binding driving forces, compounds were categorized into three groups. Dominant electrostatic contribution (e.g., CMNPD6811, CMNPD29420, CMNPD15979): characterized by multiple polar or charged moieties that engage in electrostatic interactions but also create a desolvation penalty. Combined dominance of van der Waals forces and hydrophobic interactions (e.g., CMNPD6811, CMNPD29420, CMNPD15979): a classic binding mode with shape complementarity and hydrophobic driving force, often associated with enhanced drug-likeness. Unique binding mode (e.g., CMNPD11217, CMNPD21727, CMNPD25781): positive electrostatic interaction values, with CMNPD11217 showing favorable polar solvation energy in the bound state. Hydrogen bonding contributions were generally limited. In total, 11 compounds were identified with MM/GBSA binding free energies ranging from −111.15 to −57.57 kcal/mol and |LE| ≥ 2 kcal·mol^−1^ per non-H atom.
The 11 compounds meeting |LE| ≥ 2 include benzoxepines (1), carboxylic acids and derivatives (1), harmala alkaloids (7), macrolactams (1),, and naphthofurans (1). harmala alkaloids stand out in both quantity and for having the highest binding free energy, with the top seven compounds belonging to this class. CMNPD6811 (harmala alkaloids, ΔGbind = −111.15 kcal/mol) exhibits the most favorable binding energy. Other categories include CMNPD24939 (naphthofurans, −73.87 kcal/mol), CMNPD28415 (macrolactams, −70.22 kcal/mol), CMNPD19749 (carboxylic acid derivatives, −60.67 kcal/mol), and CMNPD10150 (benzoxepines, −57.57 kcal/mol).
Mechanistically, Coulomb energy, lipophilic energy, and van der Waals energy are primary drivers for harmala alkaloids, with significantly negative ΔECoulomb values. Despite a large desolvation penalty (positiveΔGGB), the net effect remains highly favorable. Structurally, harmala alkaloids feature bulky, rigid polycyclic skeletons that engage in extensive hydrophobic interactions and π-π stacking with hydrophobic/aromatic residues (primarily phenylalanine, Phe), consistent with the findings presented in Table 4. For the other four compounds, binding is primarily driven by ΔEvdW and ΔGLipo. Hydrogen (ΔEHbond) contributions are limited for all 11 compounds, aligning with the docking results.
All 11 MNPs bind to the same active site as the positive control drug Rilematovir (Figure 9). The top six manzamine alkaloids exhibit highly consistent binding modes: the main driving force is electrostatic interactions (ΔECoulomb −120.62 to −137.26 kcal/mol); the primary antagonistic component is desolvation penalty (ΔGGB 106.20 to 138.69 kcal/mol); binding stability relies on hydrophobic interactions (ΔGLipo −40.58 to −49.61 kcal/mol) and van der Waals forces (ΔEvdW −46.70 to −64.29 kcal/mol); and hydrogen bonding (ΔEH bond) contribution is negligible. These results demonstrate that manzamine alkaloids achieve potent binding to the target pocket primarily through dominant electrostatic interactions, albeit with a substantial desolvation penalty, while their bulky, rigid scaffolds enable tight integration via hydrophobic and van der Waals contacts. In contrast, manzamine B N-oxide exhibits markedly reduced ΔECoulomb (−19.2 kcal/mol), likely due to N-oxide modification introducing steric hindrance and reducing effective positive charge, diminishing electrostatic attraction. Its ΔGGB is reduced to 57.92 kcal/mol, suggesting altered polarity and reduced desolvation penalty, though insufficient to compensate for lost electrostatic attraction. ΔGLipo and ΔEvdW remain largely unchanged, indicating that the scaffold still accesses the binding pocket, but crucial anchoring may be partially lost.
2.5. Binding Mode Interactions
Specific interactions between the 11 compounds and the F protein were explored (Figure 10). These compounds feature conjugated systems engaging in extensive hydrophobic interactions and π-stacking with the phenyl ring of Phe residues in the F protein. Additionally, heteroatoms can form hydrogen bonds with adjacent amino acid residues, such as Phe and Asp.
The detailed bond length data are presented in Table S4 in the Supplementary Materials. The hydrophobic interaction distances range from 2.0 to 4.0 Å, indicating effective hydrophobic contacts between the amino acid residues of the target protein and the ligand, which further confirms that hydrophobic interactions are a key driving force in small molecule–protein binding. The H–A distances of the hydrogen bonds are mostly in the range of 2.3–3.0 Å, indicating weak to moderate interactions and suggesting that hydrogen bonding plays a relatively minor role. Most π-stacking distances exceed 4.0 Å, indicating weak interactions with limited contribution to binding.
These compounds contain macrocyclic or complex polycyclic skeletons with multiple hydroxyl, carbonyl, or nitrogen substituents. The polycyclic skeletons of manzamine alkaloids generate significant hydrophobic, π-stacking, and van der Waals interactions, driving protein binding and conferring molecular rigidity. Flexibility from alkyl chains and rotatable bonds allows for conformational adaptation for optimal complementarity, enhancing drug-likeness. Rhytidenone A, azonazine, and spiroxin D represent another type: highly rigid and structurally compact. Rhytidenone A is planar and likely binds to the F protein via planar stacking; azonazine (bicyclo[3.3.1]nonane core) and spiroxin D (helical structure) possess fixed 3D shapes suggesting lock-and-key binding. These compounds have lower molecular weights than manzamine alkaloids. The nitrogen and oxygen atoms embedded in rigid, non-polar scaffolds contribute to moderate aqueous solubility. All these factors collectively contribute to optimal LogP and TPSA, thereby conferring favorable drug-like characteristics. Pactamide E, a macrolactam, exhibits intermediate flexibility and molecular weight, with a hydrophobic skeleton (alkyl chain) and hydrophilic functional groups (amide, hydroxyl) jointly contributing to favorable properties.
3. Discussion
In our study, potential anti-RSV molecules were screened using CADD with the CMNPD. Based on molecular docking, ADMET profiling, and MM/GBSA calculations, 11 compounds were identified as promising anti-RSV inhibitors targeting the F protein, including manzamine alkaloids (6-hydroxymanzamine A/manzamine Y, manzamine F, manzamine E, 11-hydroxymanzamine J, 8-hydroxymanzamine B, manzamine M, manzamine B N-oxide), the naphthofuran rhytidenone A, the macrolactam pactamide E, the carboxylic acid derivative azonazine, and the benzoxepine spiroxin D. All 11 compounds primarily interact with the RSV-F protein through Coulomb energy, lipophilic energy, and van der Waals energy. Molecular docking revealed superior binding affinity compared to the established F protein inhibitors Rilematovir and Sisunatovir. These compounds exhibit favorable predicted ADMET properties, indicating a high likelihood of good oral absorption and promising drug-like characteristics. The MM/GBSA-calculated binding free energies for all these compounds were highly favorable, ranging from −111.15 to −57.57 kcal/mol, indicating their predicted strong binding affinity to the target protein. However, it should be noted that the MM/GBSA approach involves several approximations (e.g., implicit solvent, neglect of entropy), which may overestimate absolute binding free energies. Thus, these calculated values are interpreted as relative ranking scores rather than absolute thermodynamic quantities [31].
Manzamine alkaloids, a unique group of β-carboline alkaloids isolated from marine sponges with broad bioactivities, such as cytotoxic, antibacterial, antifungal, antimalarial, insecticidal, anti-inflammatory, and anti-HIV activities [41], stood out due to exceptionally high binding energy and great drug-like properties. Yousaf et al. (2004) reported anti-HIV-1 activity for multiple manzamines (EC_50_ = 0.59–22.2 µM) and the first pharmacokinetic study of manzamine A, revealing absolute oral bioavailability of 20.6%, C_max_ = 1066 ng/mL, and T_max_ = 10 h, indicating good absorption and moderate oral bioavailability [42]. Palem et al. (2011) reported that manzamine A inhibited HSV-1 replication in SIRC cells at 1 µM, comparable to 50 µM acyclovir [43]. These data demonstrated broad-spectrum antiviral activity consistent with our virtual screening results.
In summary, 11 compounds, especially Manzamine alkaloids, are proposed as orally active anti-RSV drug candidates with RSV F inhibitory potential. Validating these in silico findings through in vitro and in vivo studies will be crucial for advancing these ligands as promising anti-RSV drug candidates targeting the F protein, or as starting points for further lead optimization.
However, this study has certain limitations. The current work primarily relied on conventional molecular docking as the main screening tool. While efficient for large-scale virtual screening, this static approach does not fully capture the dynamic behavior and electronic properties of the alkaloid–F protein interactions. Therefore, future studies could employ molecular dynamics simulations to assess binding stability and conformational flexibility over time, providing a more dynamic understanding of the inhibition mechanisms [44]. Additionally, PPI network analysis could be introduced to explore interactions between RSV-F protein and host factors [45], offering a system-level perspective on viral infection. Furthermore, DFT-based methods could be applied in two complementary directions: first, to validate the structural assignments of the screened alkaloids via NMR calculations [46]; and second, to investigate their electronic properties, global reactivity descriptors (such as hardness/softness), and local reactive sites (via Fukui indices) at the atomic level, thereby elucidating the intrinsic chemical reactivity that may underlie their binding affinities [47]. While these computational results provide a promising starting point for drug discovery, they need to be corroborated by subsequent in vitro experimental studies to assess their actual biological activity.
4. Materials and Methods
4.1. Docking Procedures
The chemical structures of 31,561 MNPs were obtained from the CMNPD (State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing, China). Two clinically studied F protein inhibitors, Rilematovir and Sisunatovir, were used as positive controls; their 3D structures were retrieved from the PubChem database (National Center for Biotechnology Information, U.S. National Library of Medicine, Bethesda, MD, USA). The F protein (PDB code: 5KWW) was downloaded from the Protein Data Bank (PDB) database (Rutgers, The State University of New Jersey, Piscataway, NJ, USA). Molecular docking was performed using the AutoDock 4.2.6 software package (Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA, USA), according to the standard procedure for a rigid receptor and a flexible ligand. Initial screening parameters were as follows: num_modes = 5 and exhaustiveness = 10. Subsequent docking used num_modes = 20 and exhaustiveness = 20 for accuracy. The grid size was 25.0 Å in x, y, and z directions, centered on co-crystallized ligand coordinates (18.1, 19.6, 21.1). The specified coordinates were used during the docking of each ligand. The two F protein inhibitors, Rilematovir and Sisunatovir, were used as positive controls. AutoDockTools (ADT) v.1.5.6 (Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA, USA) and PyMOL Molecular Graphics System v.3.1.6.1 (Schrödinger, LLC, New York, NY, USA) were used for preparation and visualization.
4.2. Prediction of ADMET Profile
ADMET properties (e.g., absorption, distribution, metabolism, excretion, and toxicity) were predicted using the ADMETlab 3.0 online platform [32], employing a comprehensive collection of predictive models, including machine learning algorithms and quantitative structure–activity relationship (QSAR) approaches, trained on extensively curated chemical datasets. For key parameters, the standard pre-set prediction thresholds of the platform were applied to evaluate drug-likeness and medicinal chemistry compatibility. All predictions were conducted using the default parameters as implemented in the webserver.
4.3. Binding Free Energy Calculations
Binding free energies were calculated using molecular mechanics/generalized born surface area (MM/GBSA) in Schrödinger Suite (Prime module, Maestro v.13.5.128, Schrödinger, LLC, New York, NY, USA, 2023). The calculations were performed using the OPLS4 force field and VSGB solvation model to represent the aqueous environment. All other parameters were maintained at their default settings [48].
4.4. Two-Dimensional Representation
Two-dimensional (2D) protein–ligand interaction diagrams were generated using the Ligand Interactions module of Molecular Operating Environment (MOE), v.2024.06 (Chemical Computing Group ULC, Montreal, QC, Canada).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Li Y. Wang X. Blau D.M. Caballero M.T. Feikin D.R. Gill C.J. Madhi S.A. Omer S.B. Simões E.A.F. Campbell H. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: A systematic analysis Lancet 20223992047206410.1016/S 0140-6736(22)00478-035598608 PMC 7613574 · doi ↗ · pubmed ↗
- 2Young M. Smitherman L. Socioeconomic impact of RSV hospitalization Infect. Dis. Ther.202110354510.1007/s 40121-020-00390-733656651 PMC 7926081 · doi ↗ · pubmed ↗
- 3Gatt D. Martin I. Al Fouzan R. Moraes T.J. Prevention and treatment strategies for respiratory syncytial virus (RSV)Pathogens 20231215410.3390/pathogens 1202015436839426 PMC 9961958 · doi ↗ · pubmed ↗
- 4Shi J. Huang X. Ye C. Lu Y. Liu Y. Wei Y. Wei X. Respiratory Syncytial Virus (RSV): A comprehensive overview from basic biology to clinical prevention and control Med. Res. Rev.2025 ahead of print 10.1002/med.70025 PMC 1305869141261734 · doi ↗ · pubmed ↗
- 5Keam S.J. Nirsevimab: First approval Drugs 20238318118710.1007/s 40265-022-01829-636577878 · doi ↗ · pubmed ↗
- 6Syed Y.Y. Clesrovimab: First approval Drugs 2025851487149210.1007/s 40265-025-02224-740906348 · doi ↗ · pubmed ↗
- 7Anfaal Z. Khan Z.A. Aslam M.A. FDA approves m RESVIA: Embracing the new era of RSV prevention with advanced m RNA technology Ann. Pharmacother.20255967667710.1177/1060028024130143239654133 · doi ↗ · pubmed ↗
- 8Bonneux B. Jacoby E. Ceconi M. Stobbelaar K. Delputte P. Herschke F. Direct-acting antivirals for RSV treatment, a review Antivir. Res.202422910594810.1016/j.antiviral.2024.10594838972604 · doi ↗ · pubmed ↗