From Black Box to Biological Insight: AttentioFuse Unlocks Multi-Omics Dynamics in Lung Cancer
Yuhang Huang, Yungang He, Liyan Zeng, Lei Liu, Fan Zhong

TL;DR
This paper introduces AttentioFuse, an interpretable deep learning model that improves lung cancer staging by integrating multi-omics data and providing biological insights.
Contribution
The novelty lies in the Reactome-guided mid-fusion framework that links predictions to gene-pathway evidence chains, enhancing interpretability in multi-omics models.
Findings
AttentioFuse achieves competitive performance in T- and N-stage prediction for lung cancer.
The model reveals biologically relevant pathways like AKT/mTOR and Notch signaling in lung cancer subtypes.
It provides pathway-resolved explanations that are consistent across different data folds.
Abstract
Lung cancer is often staged using imaging and pathology, but tumors within the same stage can behave very differently. Multi-omics profiles contain complementary information that may improve risk stratification, yet many deep learning models act as “black boxes” that are hard to audit clinically. We present AttentioFuse, an interpretable Reactome-guided mid-fusion framework that links predictive signals to a gene–pathway–modality evidence chain. In TCGA LUAD/LUSC cohorts, AttentioFuse achieves competitive performance for T- and N-stage prediction and provides pathway-resolved explanations that are coherent across folds. Importantly, these explanations are hypothesis-generating and do not imply causality without experimental validation. Background: Lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC), the major subtypes of non-small cell lung cancer (NSCLC), exhibit distinct…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
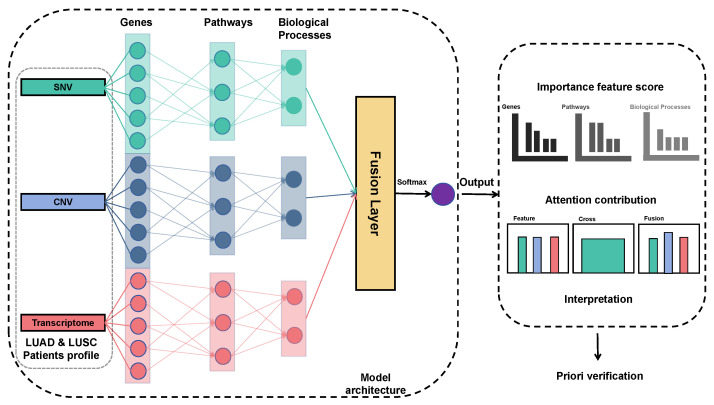 Figure 1
Figure 1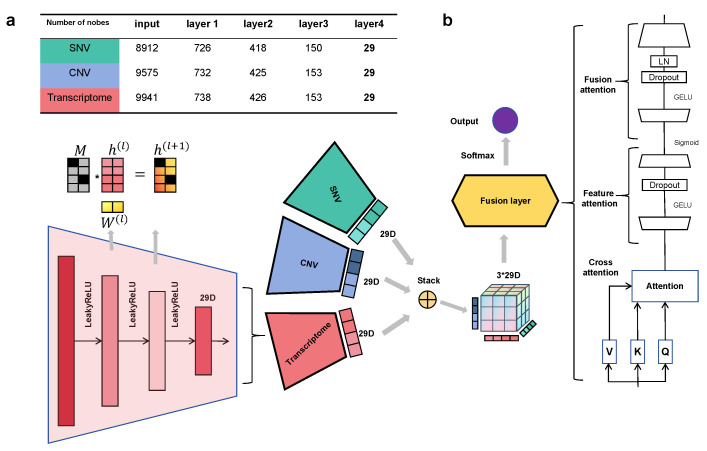 Figure 2
Figure 2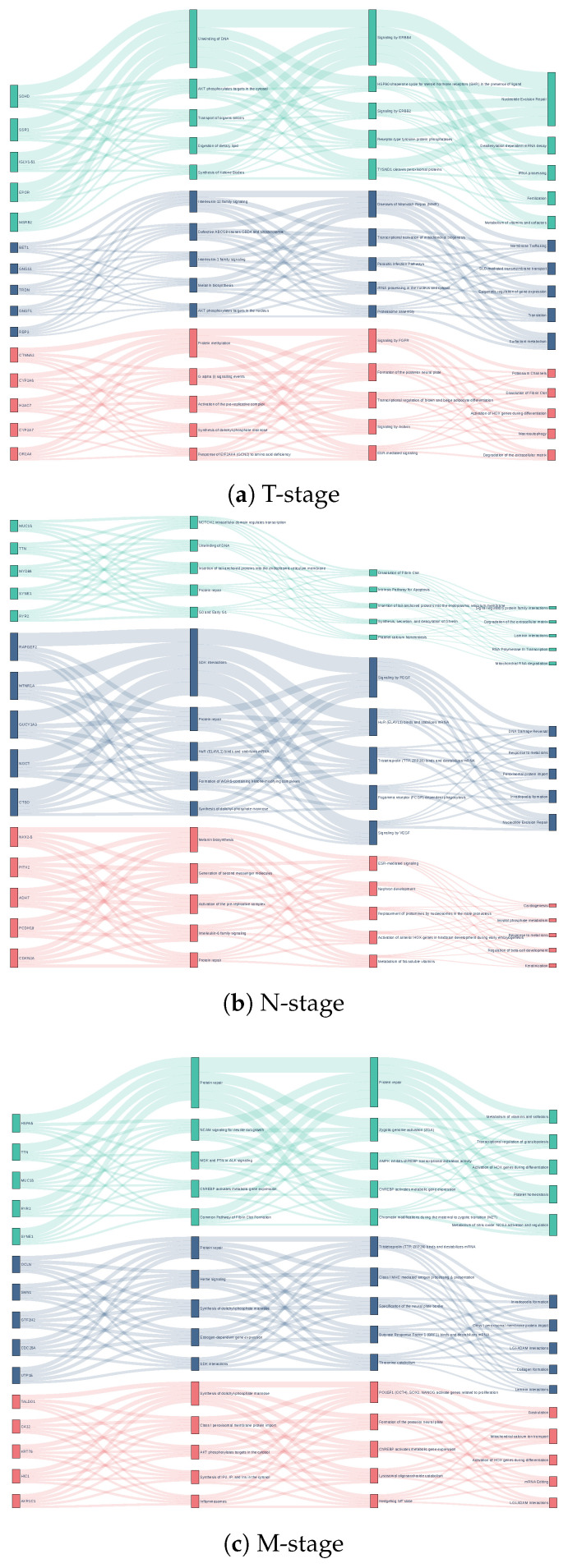 Figure 3
Figure 3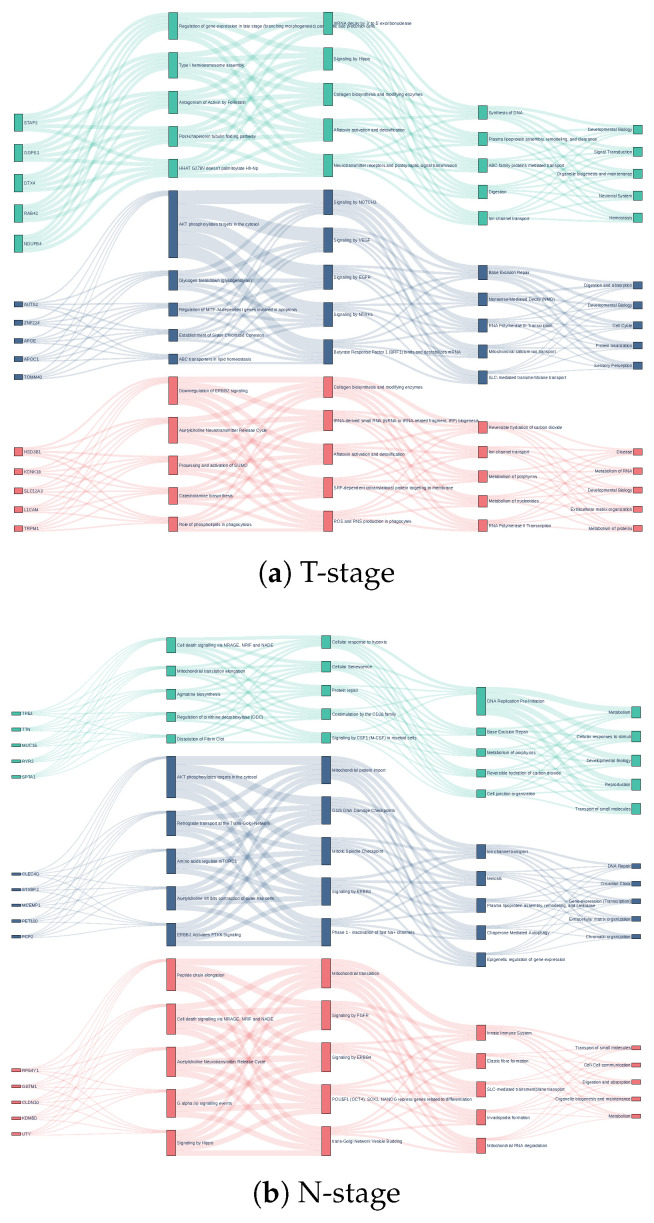 Figure 4
Figure 4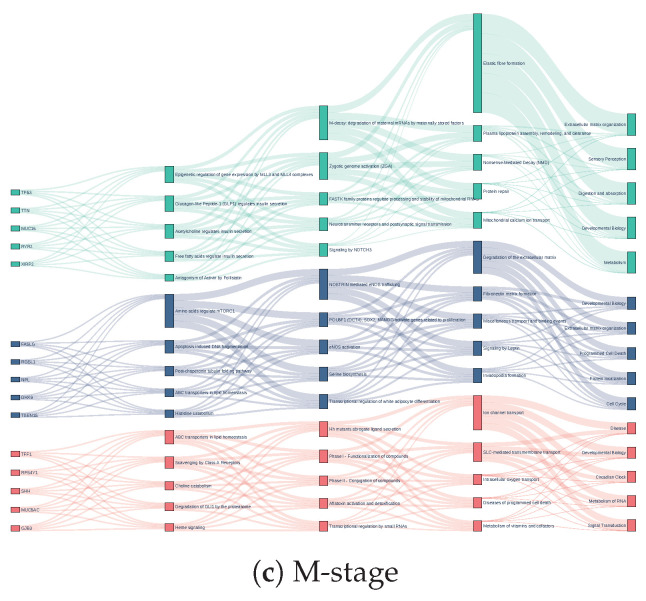 Figure 5
Figure 5- —National Key Research and Development Program of China
- —Shanghai Science and Technology Innovation Action Plan in Computational Biology
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBioinformatics and Genomic Networks · Machine Learning in Bioinformatics · Ferroptosis and cancer prognosis
1. Introduction
Non-small cell lung cancer (NSCLC) is dominated by LUAD and LUSC, two histologies that together account for over 80% of cases yet differ markedly in molecular circuitry and clinical behavior [1]. While tumor-node-metastasis (TNM) remains the clinical backbone for staging, labels are influenced by imaging/pathology workflows and can be noisy across centers, complicating the development—and interpretation—of purely molecular predictors.
Recent progress in artificial intelligence (AI), machine learning (ML), and deep learning (DL) has enabled predictive modeling from high-dimensional molecular measurements and clinical endpoints. In oncology, such models are increasingly explored for patient stratification, biomarker discovery, and treatment-related decision support, especially when informative signals are distributed across heterogeneous omics layers. However, clinical translation typically requires more than headline accuracy: models should be auditable, mechanism-linked, and robust to cohort and workflow heterogeneity [2,3,4].
Multi-omics integration (transcriptome, copy-number variation, single-nucleotide variants, etc.) is a natural route to expose mechanisms underpinning prognosis and treatment response. Deep models often achieve strong discrimination, but their black-box nature limits clinical translation where mechanism-aware evidence is required [5]. Integration strategies span early fusion (IntegratalNet; concatenate then encode), mid-fusion (independent encoders with intermediate interaction), and late/ensemble fusion (decision-level aggregation). Beyond generic fusion, biologically informed networks align hidden units to pathway hierarchies to yield pathway-level attributions (e.g., PASNet; P-NET as a mask-based variant) [6,7,8,9,10]. Parallel lines include factor/similarity approaches (MOFA, DIABLO, SNF) that learn shared/omics-specific structure [11,12,13], and recent attention-based multimodal fusion that provides modality-level weights and better tolerance to missing modalities [14,15].
Recent technical reviews provide systematic taxonomies of multi-omics integration methods, spanning classical correlation/matrix-factorization approaches to modern deep learning and deep generative architectures, and emphasize practical issues such as missing modalities, normalization, and cohort shift. These overviews motivate mid-fusion designs that preserve modality-specific encoders while explicitly modeling cross-omics interactions [16,17,18].
In parallel, there is growing emphasis on explainable AI (XAI) for omics modeling, where explanations should be reproducible across resampling or retraining and should connect feature-level signals to biologically meaningful units (e.g., pathways). Pathway-guided interpretable deep learning architectures have therefore become an active direction for reconciling predictive modeling with mechanism-oriented interpretation [19,20].
Under real-world constraints, heterogeneous cohorts pose practical issues we do not claim to eliminate: label-uncertainty in morphology-driven TNM; incomplete priors, as curation under-represents many regulatory programs; and cross-cohort heterogeneity in assay pipelines and omics availability (e.g., TCGA has SNV/CNV/transcriptome, whereas external resources may offer only a subset). Accordingly, auditability is emphasized alongside headline accuracy, and external cohorts are treated as feasibility checks under available modalities.
Although pathologic TNM is determined post-operatively, multi-omics profiles can be obtained from pre-treatment biopsies or diagnostic specimens. Our goal is therefore not to replace pathology, but to provide preoperative, mechanism-linked risk stratification that complements imaging-based staging when uncertainty exists. In the clinical timeline, the proposed prediction can be applied (i) after diagnostic sampling but before definitive therapy, to flag patients likely to harbor locally advanced disease (T3–4/N1–3) or distant spread (M1) and thus motivate intensified staging work-up and multidisciplinary planning; and (ii) perioperatively, to contextualize molecular aggressiveness and guide hypothesis-driven discussions of neoadjuvant/adjuvant strategies. Because TNM labels can be workflow-dependent and noisy across centers, we adopt clinically interpretable binarizations (Tis/T1–2 vs. T3–4; N0 vs. N1–3; M0 vs. M1) to reduce label granularity-driven variance while preserving decision-relevant thresholds.
AttentioFuse is an interpretation-oriented mid-fusion framework with (i) omics-specific encoders to preserve modality-unique signals, (ii) a hierarchical attention stack (feature-, cross-omics-, fusion-level) that yields sample-wise modality weights and pathway contributions, and (iii) integrated explainability that reconciles local DeepSHAP gene attributions with global attention summaries into a consistent gene-to-pathway-to-omics evidence chain [21]. AttentioFuse is presented in two depth variants (3F and 5X); the five-layer AttentioFuse-5X emphasizes hierarchical interpretability under end-to-end fine-tuning without materially altering model ranking. By design, the architecture remains modular when some modalities are absent (Figure 1).
Compared with prior multi-omics predictors, AttentioFuse is designed to address three gaps simultaneously: (i) preserving modality-specific structure via masked omics encoders while enabling explicit cross-omics interaction modeling at a mid-fusion stage; (ii) producing a coherent gene-to-pathway evidence chain by combining gene-level attributions with pathway-aware aggregation under Reactome priors; and (iii) offering a dual-depth instantiation under identical priors, where 3F serves as the main benchmarking backbone and 5X increases hierarchical resolution for interpretation without aiming for performance gains.
Preview on LUSC and LUAD. In LUSC-only previews, AttentioFuse-3F attains ≈0.92 accuracy (ACC)/≈0.91 F1 on T-stage and ≈0.86/≈0.85 on N-stage without class balancing, exceeding early-fusion counterparts; M-stage saturates across models at ≈0.98. In LUAD-only previews, AttentioFuse-3F reaches ≈0.91/≈0.90 on T-stage and ≈0.86–0.87 on N-stage, while M-stage is near-ceiling (≈0.93–0.97). These trends persist with or without SMOTE—balancing shifts absolute numbers slightly but does not change relative ordering [22].
Our goal is mechanism-aware decision support under realistic constraints: deliver competitive discrimination and surface pathway-resolved rationales, as well as make explicit where priors, labels, and modality availability cap performance. Alongside discrimination, we report calibration and decision-analytic summaries to contextualize utility, and we quantify explanation stability.
2. Materials and Methods
2.1. Data Acquisition and Processing
The primary cohort was TCGA LUAD/LUSC, accessed via TCGAbiolinks (downloaded September 2024) [23]. mRNA expression matrices were processed by removing mitochondrial genes (MT-*) and applying log1p; gene-level CNV followed GISTIC2, with high-confidence events retained [24]. SNVs were derived from MAF-like inputs by excluding non-functional classes (silent/UTR/intron/IGR), applying a t_alt_count > 5 filter when available (otherwise skipped with logging), deduplicating sample–gene pairs, and pivoting to a binary sample × gene mutation matrix (any qualifying variant to 1). All omics were harmonized to 12-character TCGA short IDs with duplicate aliquots averaged.
Pathologic TNM labels were binarized into clinically interpretable groups—T (Tis/T1–2 vs. T3–4), N (N0 vs. N1–3), and M (M0 vs. M1) [25]—to reduce label-granularity-driven variance and mitigate workflow-related label noise while preserving clinically actionable decision thresholds. Clinical rows were deduplicated by patient identifier; TX/NX/MX and not reported were excluded. For each analysis we aligned patients by short-ID intersection across the required modalities and re-indexed matrices accordingly. To avoid leakage, normalization and fold-wise feature-mean imputation were fit on training folds and applied to validation/test portions using training statistics only. Class imbalance was probed via an optional borderline-SMOTE (Borderline Synthetic Minority Over-sampling Technique, Borderline-SMOTE) (k = 5, capped at ≤15% synthetic samples) applied within training folds; we report results with and without this option.
External evaluation was conducted as a portability feasibility check using publicly available resources under a modality-available protocol [26,27,28]. Because external resources may differ in assay pipelines and often provide only a subset of modalities, external experiments were restricted to the modalities available in each dataset and are interpreted as generalizability signals rather than head-to-head biological comparisons. Harmonization followed consistent steps: identifier standardization and gene-name cleaning (including removal of version suffixes where applicable), feature-space alignment by intersection to a shared gene set, and within-cohort preprocessing (numeric coercion, imputation of missing values where needed, and scaling). When preprocessing parameters were transferred, transformations were fitted using training data only and applied to held-out splits without accessing external labels.
Overall, these external analyses suggest that AttentioFuse can qualitatively transfer under modality-available settings, while performance is primarily constrained by domain shift and missing modalities rather than the fusion design itself; full external portability results are provided in Supplementary Material File S1.
We assembled TCGA LUAD and LUSC multi-omics cohorts by harmonizing patient identifiers (12-character TCGA short IDs) and retaining only patients present in all required modalities. This yielded 505 LUAD and 482 LUSC patients with tri-omics profiles (mRNA, CNV, and SNV). For each staging task, we then excluded cases with missing or unknown pathologic labels (TX/NX/MX or not reported) and applied the predefined binarizations (Tis/T1–2 vs. T3–4; N0 vs. N1–3; M0 vs. M1). As a result, the final label-available sample sizes were: LUAD—T: 503 (438/65), N: 494 (326/168), M: 366 (341/25); and LUSC—T: 407 (333/74), N: 400 (263/137), M: 334 (329/5). Counts are reported as class 0/class 1.
2.2. Model Design
AttentioFuse employs mask matrices derived from the Reactome database (v86) [29] to constrain layer connectivity. We instantiate two depth variants that share the same Reactome priors: a three-layer configuration (3F) used for main classification experiments, and a five-layer configuration (5X) used when deeper pathway hierarchy and interpretation are the focus. The 5X shares identical Reactome masks and yields comparable predictive accuracy but introduces extra depth and parameters primarily useful for hierarchical interpretation (Figure 2).
2.2.1. Omics-Specific Encoders
Each molecular modality is processed through dedicated pathways using masked linear layers:
where denotes Reactome-derived binary masks, ⊙ is the Hadamard product, and is LeakyReLU with :
Intuitively, each masked layer propagates signals only along Reactome-supported gene–pathway links, so the model learns pathway-structured representations rather than unconstrained dense mappings.
Gene–pathway relationships from Reactome were encoded as binary mask matrices. During training, these masks were applied to via Hadamard product, suppressing weights of Reactome-unsupported connections and enforcing the network to learn biologically supported gene–pathway relationships.
2.2.2. Cross-Omics Attention Fusion
The fusion module processes multi-omics embeddings (m modalities; e.g., m = 3 for TCGA: mRNA/CNV/SNV) through a hierarchical attention mechanism [30]. Each modality is encoded into a 29-dimensional pathway activation vector by the Reactome-guided encoders. The multimodal integration module employs attention to dynamically weigh cross-omics interactions:
where are linear projections of the tri-omics embeddings and d is the pathway dimension for scaling.
Here, the attention weights quantify how strongly one omics embedding attends to another, enabling sample-specific cross-omics interaction modeling.
A two-layer neural gate g dynamically weights combined features:
where is the sigmoid function and stacks modality-pathway features. This gate acts as a soft feature selector, amplifying informative fused components while down-weighting less relevant ones. The final fused embedding integrates gated features while preserving original modality characteristics through residual learning:
The residual term preserves the average modality signal, which stabilizes training and avoids over-reliance on a single omics layer.
2.2.3. Interpretation Methods
For a sample x and modality m, DeepSHAP yields gene-level attributions for each input gene g (signed contributions to the logit of the positive class). We define the global gene importance as
We use the mean absolute attribution so that both positive and negative contributions are treated as evidence strength.
Let denote Reactome membership (gene g belongs to pathway node p under the masked hierarchy). We aggregate gene attributions to modality-specific pathway scores:
This step aggregates gene-level attributions to the pathway level according to Reactome membership, yielding pathway-resolved evidence within each modality.
From the fusion module we extract attention-derived modality weights , normalized by . The integrated pathway importance is then
This integrates pathway scores across modalities using attention-derived modality weights, producing a single cross-omics pathway importance.
To quantify explanation stability across folds, we compute a fold-specific pathway-importance vector and report average pairwise Spearman correlation:
A higher value indicates that the pathway ranking is more reproducible across cross-validation folds.
To increase confidence that attention-weighted explanations are not artefacts, we perform a label-permutation test: training on permuted labels should reduce predictive performance to near-chance and disrupt pathway rankings.
To quantify the additional pathway resolution enabled by the deeper masked hierarchy in AttentioFuse-5X, we summarize the attribution distribution over pathway nodes at each masked layer. Let denote the non-negative node importances for a given layer (aggregated across modalities when applicable) and the normalized attribution mass. We report (i) the number of non-negligible nodes with , and (ii) the effective number of contributing nodes . These structural metrics characterize how many pathway nodes are meaningfully resolved at deeper Reactome levels, independent of predictive performance.
These metrics summarize how many pathway nodes meaningfully carry attribution mass, providing a quantitative proxy for biological resolution.
2.3. Comparative Model Implementation
We implemented classical and neural baselines: logistic regression (L2) [31], LASSO [32], Elastic-Net [33], Gaussian Naive Bayes [34], random forest [35], XGBoost [36], and a multilayer perceptron (MLP). The MLP mirrors the hidden-layer configuration used in our models (three fully connected layers with 256 units, LeakyReLU , 30% dropout).
All experiments were evaluated using stratified 5-fold cross-validation. To prevent data leakage, preprocessing (scaling and any fold-wise imputation) was fitted on the training split of each fold only and then applied to the corresponding validation split using training-fold statistics. When enabled, Borderline-SMOTE was applied within the training split only.
We did not perform data-driven hyperparameter optimization (e.g., grid/random search) for the reported benchmarks. Instead, each model was evaluated with a fixed, pre-specified configuration or standard library defaults where appropriate, kept constant across folds and tasks (Table 1). This design avoids optimistic bias introduced by tuning on validation data and enables direct comparability across models. Accordingly, performance is reported as mean ± SD across the five folds.
2.4. Model Training Protocol
Optimization uses AdamW (Adam with decoupled weight decay) [37] (initial learning rate 0.01) with linear warm-up and Reduce-on-Plateau scheduling (factor 0.5, patience 5). Global gradient-norm clipping (L2-norm = 1.0) mitigates gradient explosions. Early stopping halts training after 10 consecutive epochs without validation loss improvement. Implementation used an NVIDIA RTX 4090 GPU with batch size 128. All linear layers were initialized with Kaiming (He) initialization [38]. Both 3F and AttentioFuse-5X are trained end-to-end under the same protocol.
3. Results
3.1. Introductory Summary of Omics-Model Characteristics
TNM staging was primarily evaluated on SMOTE-balanced datasets with oversampling confined to training folds inside stratified 5-fold CV. Accuracy (ACC) and weighted F1-score (F1) were used as headline metrics. The 3F model served as the main predictive backbone, whereas the 5X configuration was reserved for hierarchical interpretation. Under the balanced protocol, 3F and 5X exhibited highly similar trends (Supplementary Material File S2).
Under SMOTE balancing, M-stage classification reached near-ceiling performance across models. The near-ceiling performance for M-stage (especially in LUSC) is consistent with strong class separability and/or limited label complexity under the adopted M0 vs. M1 binarization. In TCGA, M1 cases are comparatively rare, and oversampling within training folds can further simplify discrimination by amplifying a small set of highly predictive molecular signatures. As a result, multiple model families achieve similarly high ACC/F1 and M-stage becomes less informative for comparing model capacity. We therefore focus mechanistic interpretation primarily on T- and N-stages, where discrimination is more challenging and pathway-level explanations are more diagnostic. For T-stage and N-stage, AttentioFuse-3F consistently matched or outperformed neural baselines and tree/boosting methods (Table 2). To mitigate concerns about oversampling artefacts, 3F without SMOTE was also evaluated under the same CV protocol; observed changes were small and task-dependent, supporting the SMOTE-balanced ACC/F1 as primary readouts. All metrics are summarized as mean ± SD across folds to reflect variability and robustness.
3.2. Quantitative Comparison Between 3F and 5X
To support the positioning of AttentioFuse-5X as an interpretation-oriented variant, we compared 3F and 5X in predictive quality under the same task definitions. The 3F configuration is reported with 5-fold cross-validation (mean ± SD; Table 2) as our main benchmarking backbone. The 5X configuration was evaluated in a dedicated interpretability-oriented setting to verify that additional depth does not materially degrade discrimination, while enabling deeper pathway-level analyses. As summarized in Table 3, 5X achieves comparable F1-scores on both LUAD and LUSC across TNM endpoints, supporting its use primarily for hierarchical interpretation rather than performance gains.
Beyond predictive performance (Table 3), we quantified the biological resolution enabled by the additional masked hierarchy in AttentioFuse-5X using the pathway-node attribution distributions underlying the Sankey visualizations (Supplementary Material File S3). In the two deeper layers unique to 5X, the intermediate layer (Layer 3; 153 nodes) resolved 50–110 non-negligible pathway nodes across tasks, with ranging from 11.30 to 85.97, while the top pathway layer (Layer 4; 29 nodes) retained 19–29 non-negligible nodes with ranging from 4.54 to 11.27. These results support that 5X provides additional hierarchical pathway structure at deeper Reactome levels, whereas 3F cannot expose these levels by design.
3.3. Interpretability Analysis of LUAD and LUSC Staging Characteristics
Unless otherwise noted, interpretability analyses were conducted with the five-layer AttentioFuse-5X variant, while predictive benchmarking relied on the 3F configuration. Attention maps and Sankey diagrams were derived by aggregating DeepSHAP gene attributions with global attention weights across cross-validation folds; pathway-level scores were normalized within stage.
3.3.1. Validated Pathways and Novel Mechanisms in LUSC
The framework demonstrated robust alignment with canonical squamous carcinoma biology while proposing testable hypotheses for LUSC progression (Figure 3). Key validated mechanisms included Hippo–Notch signaling crosstalk (T-stage), PDGFR/VEGF-linked metalloregulation (N-stage) [39], and ERBB–HSP90 functional coupling (M-stage), consistent with established LUSC molecular programs and therapeutic targets.
Putative novel signals centered on developmental pathway reactivation [40] (including germ-layer formation programs) and microbiota-associated metastasis [41], with signatures suggestive of parasite-infection–mitochondrial crosstalk. An unexpected melanogenesis signal [42] may reflect ROS regulation beyond pigmentation. In addition, WDR5–FCGR epigenetic linkages implied macrophage polarization via histone modification [43], warranting experimental validation.
To quantify the pathway-level structure summarized in Figure 3, we analyzed the normalized attribution distribution over the top Reactome pathway layer (Layer 4; 29 nodes) that underlies the Sankey visualization (Supplementary Material File S3). In LUSC T-stage, the top five pathways account for 78.7% of the attribution mass (top-10: 93.6%), dominated by Hemostasis (22.3%), Signal Transduction (17.9%), and Cell Cycle (16.3%). In N-stage, the top five account for 71.9% (top-10: 89.3%), led by Digestion and Absorption (27.6%) and Hemostasis (20.9%). In M-stage, the top five account for 69.3% (top-10: 89.2%), with Developmental Biology (21.6%) and Circadian Clock (18.9%) among the dominant contributors. Consistently, the effective number of contributing pathways at Layer 4 remains non-trivial (T: , N: , M: ), indicating a concentrated yet multi-module explanatory structure rather than diffuse, low-specificity attribution.
3.3.2. Validated Pathways and Novel Mechanisms in LUAD
For adenocarcinoma, the model recapitulated mTORC1-mediated metabolic control in T-stage [44] and AKT–VEGF-linked angiogenic coupling in M-stage [45], aligning with LUAD metabolic and vascular dependencies. Putative novel hypotheses included embryonic nonsense-mediated mRNA decay activation [46] potentially sustaining stemness, and leptin signaling [47] consistent with obesity-associated matrix remodeling.
Mitochondrial calcium–FASTK interactions [48] suggested ion-flux-regulated RNA stability as a metabolic plasticity mechanism. Metastatic programs further implicated non-canonical collagen–Hedgehog crosstalk [49] linking extracellular mechanics to post-translational regulation. Finally, tRNA-derived networks [50] supported a potential epigenetic–metastatic coupling axis, consistent with TIMP3-linked invasion control (Figure 4).
We similarly quantified the pathway-level attribution structure summarized in Figure 4 using the normalized Layer 4 distribution (29 nodes; Supplementary Material File S3). LUAD T-stage shows a highly concentrated profile, with the top five pathways explaining 96.1% of the mass (top-10: 97.9%), dominated by Developmental Biology (49.8%) and Extracellular Matrix Organization (21.1%). In contrast, LUAD N- and M-stages exhibit broader multi-module support: for N-stage, the top five pathways account for 73.0% (top-10: 88.8%), led by Metabolism (40.6%); for M-stage, the top five pathways account for 77.2% (top-10: 91.5%), with Developmental Biology (34.0%), Disease (16.4%), and Signal Transduction (13.4%) among the leading contributors. The corresponding effective numbers further reflect this stage dependence (T: vs. N: and M: ), providing a quantitative basis for the differing degrees of pathway concentration observed in the Sankey summaries.
3.4. NSCLC Common Mechanisms and Personalized Therapeutic Implications
Analyses revealed conserved oncogenic circuitry across LUAD and LUSC while highlighting histology-specific vulnerabilities. The AKT/mTOR axis emerged as a pan-NSCLC regulator across stages. Notch signaling exhibited histology-divergent roles; -secretase inhibitors combined with anti-angiogenics may warrant hypothesis-driven evaluation. Shared extracellular matrix remodeling signatures suggested potential liquid biopsy correlates. These histology-specific differences are summarized in Table 4, while modality-specific attention contributions are reported in Table 5; the full cross-omics, feature-level, and fusion-layer attention analyses are provided in Supplementary Material Figure S1.
4. Discussion
AttentioFuse demonstrates that model transparency can be achieved without sacrificing predictive performance in NSCLC multi-omics analysis. With Reactome-constrained encoders and a hierarchical attention stack, attributions are read coherently across levels—genes, pathways, and modalities—using DeepSHAP-based gradients. The 3F variant remains the primary predictive backbone due to its favorable bias–variance trade-off, while the 5X variant provides deeper pathway resolution with similar discrimination.
Several limitations should be considered when interpreting the present findings. First, interpretability is bounded by pathway prior coverage and curation quality; signals outside Reactome annotations may be under-represented. Second, TNM labels are workflow-dependent and may contain noise; our binarized endpoints reduce granularity-driven variance but cannot eliminate label uncertainty. Third, cohort size and class imbalance (notably for M1) can lead to near-ceiling performance and reduces the value of M-stage for model comparison. Fourth, attention gates and linear masks mainly capture approximately linear interactions within each attention slice, potentially under-modeling higher-order effects when modalities are sparse or missing. Finally, external evaluations are feasibility checks under modality-available settings; domain shift across cohorts (assay pipelines, cohort composition, and missing modalities) limits direct comparability and motivates future prospective validation on clinically collected multi-omics biopsies with harmonized protocols.
Importantly, the proposed explanations are hypothesis-generating: they highlight associations consistent with pathway priors and model attributions but do not imply causal effects without experimental validation.
From a translational perspective, AttentioFuse is intended for pre-treatment settings where diagnostic biopsies can be profiled (e.g., targeted sequencing and transcriptomics) to complement imaging-based staging. The model output can serve as a decision-support signal to prioritize intensified staging work-up (e.g., additional imaging or nodal assessment), support multidisciplinary planning, and provide pathway-resolved hypotheses that may guide biomarker-focused discussion of neoadjuvant/adjuvant strategies. The framework is not a replacement for pathology; rather, it provides mechanism-linked risk stratification that can be audited and interpreted alongside clinical findings.
5. Conclusions
AttentioFuse provides an interpretable, Reactome-guided mid-fusion framework for multi-omics staging prediction in LUAD and LUSC. By combining pathway-masked encoders with hierarchical attention and DeepSHAP-based attribution, the framework links predictive signals to a coherent gene–pathway–modality evidence chain.
Across TCGA cohorts, AttentioFuse achieves competitive discrimination for T- and N-stage and near-ceiling performance for M-stage under a binarized endpoint definition. The dual-depth design enables practical trade-offs: the 3F configuration serves as a robust benchmarking backbone, whereas the 5X configuration increases hierarchical resolution for interpretation while maintaining comparable predictive quality.
Future work should focus on prospective validation in clinically collected biopsy cohorts with harmonized pipelines and on extending the modality-available protocol to broader real-world settings. Importantly, the reported pathway findings are hypothesis-generating and are intended to prioritize mechanistic follow-up rather than imply causal inference.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alduais Y. Non-small cell lung cancer (NSCLC): A review of risk factors, diagnosis, and treatment Medicine 2023102 e 3289910.1097/MD.000000000003289936827002 PMC 11309591 · doi ↗ · pubmed ↗
- 2Yates J. Van Allen E.M. New horizons at the interface of artificial intelligence and translational cancer research Cancer Cell 20254370872710.1016/j.ccell.2025.03.01840233719 PMC 12007700 · doi ↗ · pubmed ↗
- 3Lotter W. Hassett M. Schultz N. Kehl K. Van Allen E. Cerami E. Artificial Intelligence in Oncology: Current Landscape, Challenges, and Future Directions Cancer Discov.20241471172610.1158/2159-8290.CD-23-119938597966 PMC 11131133 · doi ↗ · pubmed ↗
- 4Huang D. Li Z. Jiang T. Yang C. Li N. Artificial intelligence in lung cancer: Current applications, future perspectives, and challenges Front. Oncol.202414148631010.3389/fonc.2024.148631039763611 PMC 11700796 · doi ↗ · pubmed ↗
- 5Wysocka M. A systematic review of biologically-informed deep learning models for cancer: Fundamental trends for encoding and interpreting oncology data BMC Bioinform.20232419810.1186/s 12859-023-05262-8PMC 1018665837189058 · doi ↗ · pubmed ↗
- 6Elmarakeby H.A. Hwang D. Arafeh R. Crowdis J. Gang S. Liu D. Al Dubayan S.H. Salari K. Kregel S. Richter C. Biologically informed deep neural network for prostate cancer discovery Nature 202159834835210.1038/s 41586-021-03922-434552244 PMC 8514339 · doi ↗ · pubmed ↗
- 7van Hilten A. Kushner S.A. Kayser M. Ikram M.A. Adams H.H.H. Klaver C.C.W. Niessen W.J. Roshchupkin G.V. Gen Net framework: Interpretable deep learning for predicting phenotypes from genetic data Commun. Biol.20214109410.1038/s 42003-021-02622-z 34535759 PMC 8448759 · doi ↗ · pubmed ↗
- 8Ghosh Roy G. Geard N. Verspoor K. He S. MPVNN: Mutated Pathway Visible Neural Network architecture for interpretable prediction of cancer-specific survival risk Bioinformatics 2022385026503210.1093/bioinformatics/btac 63636124954 · doi ↗ · pubmed ↗