Icariin Against Neurodegeneration: A Focus in Cell Death Pathways
Vinícius Rodrigues-Soares, Heitor Roque da Cruz, Gabriel Ferreira dos Santos, João Gabriel Restier, Pedro Castilho, Natan Giovanni Ferreira dos Santos, Elias Avgerino dos Santos, Daniel Souza Monteiro de Araújo, Karin da Costa Calaza, Rafael Brito

TL;DR
This review explores how the flavonoid icariin may protect against neurodegeneration by targeting specific cell death pathways.
Contribution
The paper provides a synthesis of icariin's molecular mechanisms in modulating distinct cell death pathways in neurological models.
Findings
Icariin shows potential to intercept apoptosis, ferroptosis, and excitotoxicity.
Current evidence on icariin's interaction with specific cell death mechanisms is fragmented.
The review clarifies icariin's molecular logic in modulating neurological cell death pathways.
Abstract
Distinct molecular pathways of cell death drive the progression of neurological pathologies. Targeting the signaling cascades induced by oxidative stress and inflammation within these pathways is central to developing cytoprotective therapies against apoptosis, ferroptosis, and excitotoxicity. In recent years, the flavonoid icariin has emerged as a promising candidate, showing significant potential to intercept these specific cell death mechanisms. However, while the current literature highlights icariin’s general neuroprotective effects, evidence regarding its precise interaction with distinct cell death modalities remains fragmented. This review synthesizes current knowledge to clarify the molecular logic by which icariin modulates these specific pathways in neurological models.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
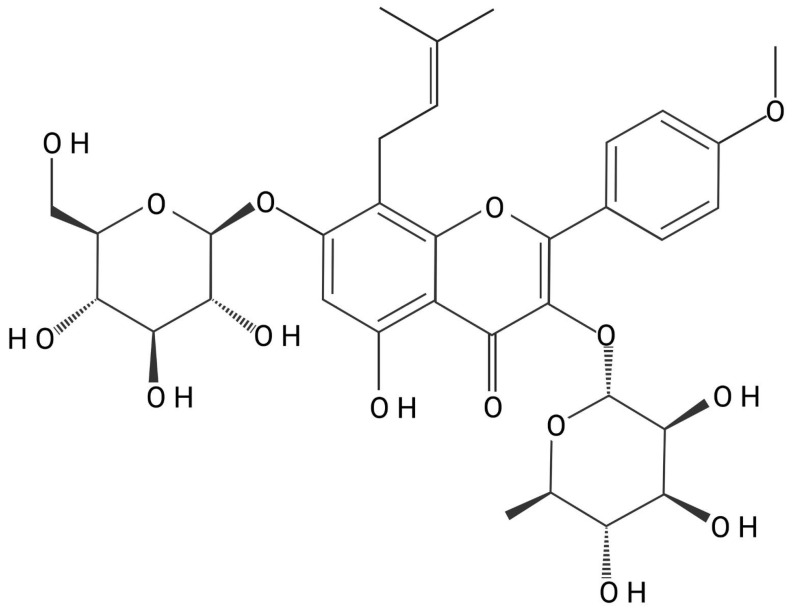 Figure 1
Figure 1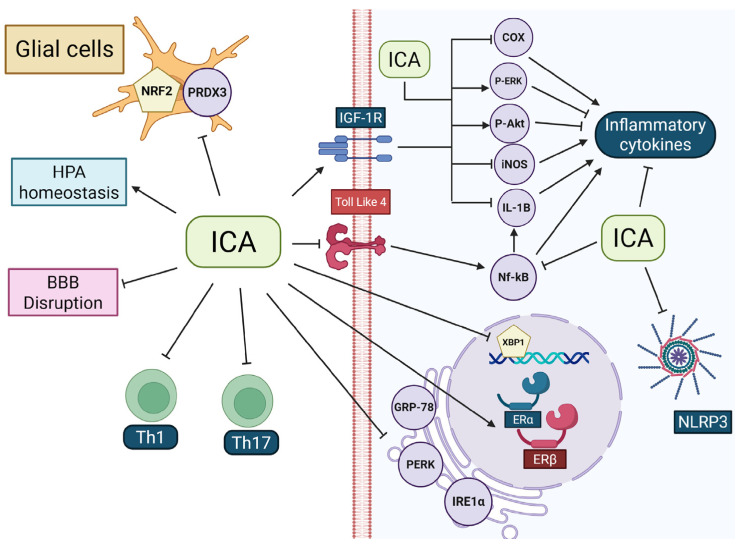 Figure 2
Figure 2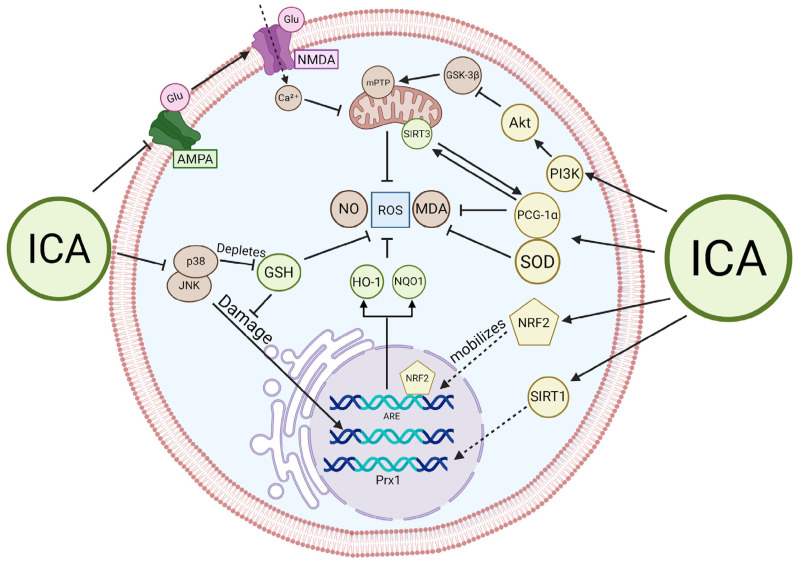 Figure 3
Figure 3 Figure 4
Figure 4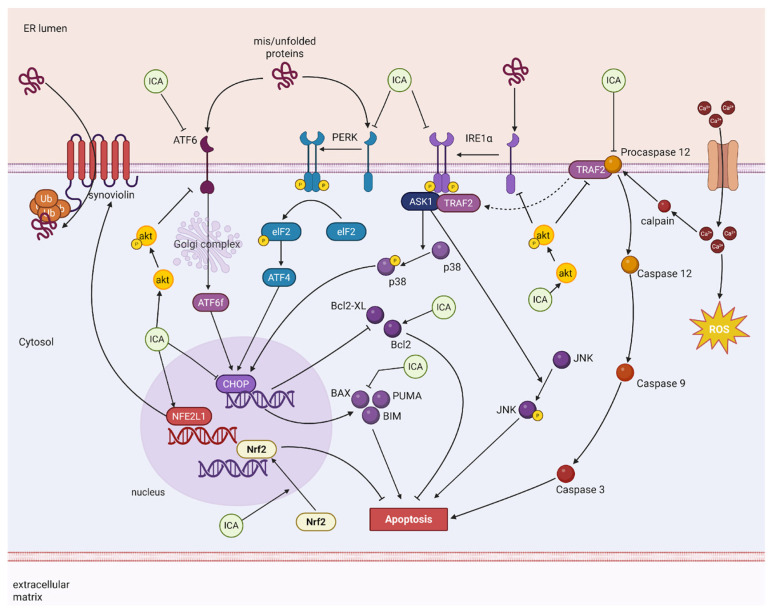 Figure 5
Figure 5- —National Council of Scientific and Technological Development (CNPq), Universal
- —National Institute of Science and Technology— INCT da GLIA (INCT/IGlia)
- —Programa Conhecimento Brasil–Atração e Fixação de Talentos
- —Fellowships from the Coordination of Superior Level Staff Improvement (CAPES)
- —Foundation for Research Support of the State of Rio de Janeiro (FAPERJ)
- —special scholarship for senior scientists of the Rio de Janeiro State
- —CNPq
- —special scholarship BCB-1
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMedicinal Plant Pharmacodynamics Research · Studies on Chitinases and Chitosanases · GDF15 and Related Biomarkers
1. Introduction
The aging of the population has been accompanied by a rising incidence of age-related and metabolic disorders, such as Parkinson’s disease, Alzheimer’s disease, age-related macular degeneration, and type 2 diabetes [1]. These conditions place a substantial economic burden on health care systems and highlight the urgent need for innovative therapeutic strategies focused not only on treatment but also on prevention [2,3]. However, the therapeutic management of neurodegenerative disorders remains a critical pharmacological challenge, largely due to the multifactorial nature of neuronal cell death. Effective intervention requires agents capable not only of crossing the blood–brain barrier but also of simultaneously modulating distinct deleterious signaling cascades that drive disease progression.
Neuronal loss and the cognitive deficits associated with neurodegenerative pathologies involve events related to inflammation, oxidative stress, and apoptosis [4,5,6,7,8]. Aberrant redox signaling in oxidative stress damages organelles, proteins, lipids, and DNA; this cellular damage is consistent with post-mortem findings in patients with Alzheimer’s, Parkinson’s and Huntington’s disease [9]. Oxidative stress can trigger specific cell death pathways, including apoptosis and ferroptosis. In apoptosis, mitochondrial membrane damage leads to a decrease in membrane potential, promoting the intrinsic cascade, a known marker in major neurodegenerative diseases such as Alzheimer, Parkinson, and amyotrophic lateral sclerosis [10]. Conversely, ferroptosis is driven by hydroperoxides initiating chain reactions facilitated by transition metals like iron. This results in the generation of highly reactive lipid radicals, a process closely linked to Parkinson’s disease [11]. On the other hand, neuroinflammation creates an environment favorable for cell death [12]. The inflammatory response varies according to pathology, ranging from microglial and glial activation in Alzheimer’s disease to leukocyte migration in stroke or amyotrophic lateral sclerosis [13]. Chronic glial activation can drive both oxidative stress and inflammatory cell death, such as pyroptosis [13]. Other signaling pathways, such as endoplasmic reticulum stress, also promote neurodegeneration and interplay with apoptosis to drive Parkinson’s and Alzheimer’s disease progression [14,15]. Moreover, lysosomal dysfunction impairs the autophagy-dependent recycling of macronutrients and organelles, further contributing to neuronal death in multiple neurodegenerative diseases [16,17]. In this context, prenylated flavonoids have emerged as privileged scaffolds for neurodrug discovery due to their favorable lipophilicity and multimodal activity [18].
Traditional medicine provides a valuable repository of bioactive compounds derived from herbs and roots that have been used for millennia to treat a wide variety of diseases. A well-known example is Herba Epimedii, widely used in traditional Chinese medicine for treating headaches, muscle weakness, and contracture in countries such as China, Korea, and Japan [19]. Icariin (ICA, C33H40O15) (Figure 1), the major bioactive constituent of Herba Epimedii (Epimedium spp.), represents a prime flavonoid candidate [20]. ICA, first identified in 1990 through chromatographic and spectroscopic methods, belongs to the class of prenylated flavonoids [21]. Despite its traditional use, the clinical translation of ICA in Western markets faces significant regulatory hurdles. A recent analysis of the European market highlights that Epimedium species are frequently found in products lacking proper authorization [22]. Unlike widespread botanicals such as Ginseng, Epimedium is currently absent from many recommendation documents worldwide, confining it to a borderline status between food supplement and medicine. To bridge the gap from an unregulated botanical to a targeted therapeutic, a precise understanding of its molecular mechanisms could facilitate translation [22].
Preclinical studies have shown that ICA exerts a wide spectrum of biological effects, including antioxidant, antitumor, anti-inflammatory, and protective activities [23]. In the central nervous system (CNS), ICA has been reported to counteract neuronal apoptosis, attenuate cognitive decline in experimental models of Parkinson’s and Alzheimer’s disease [24,25], and protect against diverse neuronal insults [24]. These beneficial effects are associated with its polyphenolic nature, which enables redox regulation by balancing reactive oxygen (ROS) and reactive nitrogen species (RNS) [24,26]. Beyond its antioxidant properties, recent evidence suggests that ICA’s neuroprotective potential also involves the release of neurotrophic factors [27,28], activation of pro-survival signaling pathways [24,29], and modulation of sirtuin family proteins, key regulators of cellular metabolism and stress response [30,31,32]. Moreover, ICA has been reported to inhibit apoptosis by reducing endoplasmic reticulum stress [24,31,33], suppressing glial reactivity [24,34], and restoring autophagic activity under pathological conditions [24,35]. Importantly, it can also interfere with ferroptosis, an iron-dependent and caspase-independent mode of regulated cell death [36].
The effects observed with ICA are consistent with findings reported for other flavonoids. Prunin, for example, has been shown to modulate several cellular pathways, presenting differently in healthy versus cancerous cells [37]. Many flavonoids can alleviate inflammation and oxidative stress, as well as activate apoptotic processes in tumors (anti-cancerous) while inhibiting it in healthy cells [38,39,40]. Although the anti-inflammatory mechanisms of polyphenolic flavonoids are not completely elucidated, their activity has been correlated with the inhibition of inflammatory signaling and reductions in proinflammatory mediators [41,42]. The antioxidative effects of flavonoids, on the other hand, have been linked to their role as ROS scavengers, lowered mitochondrial ROS production, and effective activation of nuclear factor erythroid 2-related factor 2 (Nrf2) [43,44,45,46]. As natural flavonoids, the actions of ICA are expected to present fewer negative side effects [37,47,48]. Taken together, these findings highlight ICA as a promising natural compound with multimodal actions in the CNS. This review synthesizes fragmented preclinical evidence to map these specific molecular targets (Supplementary Materials), providing the mechanistic validation necessary to support its future therapeutic standardization.
Pharmacokinetics of Icariin
The pharmacokinetic profile of ICA represents a critical determinant of its biological activity and constitutes a major source of variability across experimental models. Importantly, pharmacokinetic constraints impose limitations on the interpretation of ICA’s neuroprotective effects, particularly in studies addressing CNS outcomes. ICA exhibits low oral bioavailability due to its diglycosylated structure, which limits direct intestinal absorption [20,49,50]. Following oral administration, ICA undergoes rapid systemic clearance, with reported half-lives ranging from approximately 1 to 3 h depending on the route of administration [20,49,50]. Intravenous delivery results in a rapid decline in plasma concentrations, with a half-life of approximately 0.5 h [51], whereas intragastric administration yields bioavailability values close to 12% and a half-life of approximately 1 h [52,53]. These data consistently indicate that ICA is rapidly eliminated from systemic circulation.
Tissue distribution studies indicate that ICA accumulates preferentially in peripheral organs such as the liver, lung, spleen, heart, kidney, brain and reproductive tissues, while its direct penetration into brain tissue appears limited [50]. This restricted CNS exposure suggests that the neuroprotective effects observed in preclinical models may not solely depend on high concentrations of the parent compound within brain parenchyma. Instead, these effects are likely shaped by the contribution of bioactive metabolites, indirect modulation of peripheral immune and inflammatory signaling, and cumulative low-level exposure capable of influencing glial and redox-sensitive pathways in the CNS.
A defining feature of ICA pharmacokinetics is its extensive intestinal and hepatic metabolism. Following oral administration, ICA is predominantly converted into bioactive metabolites, including icariside I, icariside II, and icaritin, through the combined actions of intestinal enzymes, gut microbiota β-glucosidases, and efflux transporters such as P-glycoprotein [49,54,55,56,57]. Notably, the intestine represents the primary site of ICA biotransformation, with approximately 90% of the orally administered compound metabolized prior to systemic circulation, whereas intravenous administration results in minimal conversion [58,59]. These metabolites can subsequently undergo conjugation reactions, generating glucuronidated derivatives such as icaritin-7-O-glucuronide and icaritin-3-O-rhamnoside-7-O-glucuronide, which may exhibit distinct pharmacodynamic properties [57]. This metabolic complexity has important implications for the interpretation of experimental findings. The reported neuroprotective effects attributed to ICA may reflect, at least in part, the actions of its metabolites rather than the parent molecule itself. Moreover, differences in dose, route of administration, formulation, and treatment duration across studies likely represent compensatory strategies to overcome rapid clearance and limited brain distribution, contributing to heterogeneity in the molecular pathways reported to be modulated by ICA.
Several formulation-based approaches have been explored to improve ICA bioavailability, including complexation with hydroxypropyl-β-cyclodextrin and liposomal encapsulation in propylene glycol systems [54,60]. These strategies increase systemic exposure, tissue distribution, and area under the curve, nearly doubling peak plasma concentrations in some models [60]. However, improvements in brain penetration remain modest, with increases of approximately 2%, and clearance rates largely unchanged [54,60]. These findings reinforce the notion that ICA-mediated CNS effects may involve indirect mechanisms, such as modulation of peripheral inflammation, systemic oxidative stress, or sustained low-level exposure rather than acute high brain concentrations.
While pharmacokinetic data consistently indicate rapid degradation and low oral bioavailability of ICA [50], the definition of an effective dose is also dependent on the target tissue and its associated biological barriers. Recent studies reveal a marked dose–response stratification between central and non-central targets, underscoring the impact of tissue distribution on pharmacological efficacy. For example, neuroprotection in models involving CNS dysfunction required relatively high oral doses (up to 100 mg/kg), as reported in models of encephalopathy-associated neural injury [61], whereas significant cytoprotection in ocular tissues, such as prevention of lens opacification, was achieved at substantially lower doses (20 mg/kg) [62]. This discrepancy suggests that, although ICA exerts potent biological effects at low systemic concentrations, achieving efficacy in CNS-related models likely requires higher doses, reflecting limitations in neuronal tissue exposure rather than intrinsic pharmacodynamic inefficacy. Accordingly, variability in effective doses across experimental models should be interpreted in light of pharmacokinetic constraints, including clearance rate, metabolic conversion, and tissue accessibility, rather than as inconsistencies in molecular mechanisms.
Altogether, pharmacokinetic data of ICA reveal rapid degradation, low oral bioavailability, and limited distribution to neural tissues [50]. Nevertheless, numerous studies employing oral administration of ICA in animal models consistently demonstrate neuroprotective effects, including the attenuation of inflammation [34,63], oxidative stress [64,65], and apoptosis [66,67], suggesting that effective biological activity can be achieved despite pharmacokinetic constraints when appropriate dose regimens are applied.
2. Anti-Inflammatory Actions of ICA
Inflammation is a fundamental biological response of the immune system to harmful stimuli such as pathogens, damaged cells, or irritants. In its acute form, it is a short-lived, localized process essential for defense and tissue repair, allowing for the elimination of the cause of injury and initiating recovery [68]. However, dysregulated or persistent inflammation contributes to disease onset and progression, including neurodegenerative disorders, by disrupting cytokine signaling, intracellular pathways, and glial homeostasis [68]. The inflammatory response can also originate from the nervous tissue itself, a response of glial cells in pathological conditions that is called neuroinflammation. Understanding the molecular and cellular mechanisms that govern inflammatory response is therefore crucial for developing therapeutic strategies against neurodegeneration and restoring homeostasis.
ICA has shown promising therapeutic potential in neurodegenerative conditions, particularly in settings where immune-mediated inflammation is a key driver of progressive neuronal and glial degeneration. Wei and coworkers (2016) demonstrated that ICA exerts estrogen-like effects in experimental autoimmune encephalomyelitis, a chronic demyelinating inflammatory disease that promotes neurodegeneration in multiple sclerosis, through modulation of estrogen receptor alpha and beta (ERα/ERβ) expression, contributing to preservation of cerebral white matter and attenuation of secondary neuronal damage [69]. At the level of the central nervous system, Cong et al. (2020) reported that ICA suppresses microglial reactivity and downregulates proinflammatory mediators, including tumor necrosis factor alpha (TNF-α) and inducible nitric oxide synthase (iNOS), through inhibition of nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) signaling, further limiting inflammation-driven neurodegeneration in encephalomyelitis [70]. Supporting these findings, Wang et al. (2022) demonstrated that ICA attenuates glial-mediated inflammation in a C57BL/6 mouse model of autoimmune uveitis, a condition capable of inducing secondary neurodegenerative changes in central nervous system structures such as the retina [71]. In this context, ICA shifted microglial activity toward a less reactive, tissue-protective profile via peroxiredoxin-3 (PRDX3), a mitochondrial enzyme involved in hydrogen peroxide (H_2_O_2_) detoxification, whose silencing abolished ICA’s anti-inflammatory effects [71]. Consistently, Shen et al. (2015) showed that ICA induces systemic immune modulation in the same model by suppressing T helper 1 (Th1) and T helper 17 (Th17) differentiation, which was associated with reduced blood–brain barrier (BBB) permeability and improved clinical outcomes [72]. Corroborating with these findings, Pokkula and Thakur (2021) demonstrated that oral ICA administration in Wistar rats subjected to a sciatic nerve injury reduced proinflammatory cytokines, Interleucin-6 (IL-6) and TNF-α, in sciatic nerve homogenates [73]. These molecular changes were accompanied by a significant reduction in pain scores. Further, ICA-loaded nanofibers reduced inflammation markers (TNF-α, Hypoxia-Inducible Factor 1 Alpha [HIF-1α], IL-6, and Interleukin 1-β [IL-1β]) in rat sciatic nerve homogenates [74].
In models of cognitive impairment and neurodegenerative disease, ICA consistently suppresses inflammatory pathways that exacerbate neuronal loss and synaptic dysfunction. In postoperative cognitive dysfunction and Alzheimer’s disease models, ICA improves cognitive performance by reducing hippocampal inflammation, inhibiting microglial activation, and limiting neuronal injury through suppression of Toll-like receptor 4 (TLR4)/NF-κB signaling [75]. Additionally, ICA inhibits cGAS–STING signaling and its downstream effectors TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), thereby reducing type I interferon production and chronic inflammatory amplification, processes closely linked to neurodegenerative progression in Alzheimer’s disease [76]. ICA further limits disease pathology by reducing β-amyloid plaque accumulation, transforming growth factor beta 1 (TGF-β1) expression, ceramide levels, and microglial pyroptosis via downregulation of the cyclooxygenase-2 (COX-2)–NLRP3 inflammasome–gasdermin D axis [75,76,77,78]. A similar mechanistic link between inflammation and neurodegeneration is evident in epilepsy, where recurrent seizures promote excitotoxicity, oxidative stress, and chronic microglial activation. In this context, ICA reduces seizure severity and mortality during the acute phase and improves cognitive performance during the chronic phase by promoting microglial polarization toward an anti-inflammatory phenotype, thereby limiting inflammation-associated neurodegenerative damage [79]. ICA also exerts robust neuroprotective effects in ischemic and vascular-related neurodegeneration. In cerebral ischemia and vascular dementia models, where hypoxia-induced inflammation and oxidative stress drive progressive neuronal loss, ICA improves neurological outcomes, reduces cerebral edema, and suppresses microglial activation and proinflammatory cytokine production [34,80]. These anti-inflammatory effects are accompanied by restoration of synaptic and neurotrophic signaling, including increased brain-derived neurotrophic factor (BDNF), tropomyosin receptor kinase B, extracellular signal-regulated kinases (ERK), cAMP response element-binding protein (CREB), synaptophysin, postsynaptic density protein 95 (PSD-95), glutamate ionotropic receptor NMDA type subunit 2B (GluN2B), and calcium/calmodulin-dependent protein kinase II (CaMKII) expression, supporting neuronal survival and synaptic plasticity in degenerative contexts [81].
The anti-inflammatory and neuroprotective effects of ICA are, in several contexts, mediated by activation of nuclear factor erythroid 2–related factor 2 (Nrf2), a transcription factor critically involved in cellular survival and redox homeostasis. In BV-2 microglial cells, ICA suppressed lipopolysaccharide-induced production of proinflammatory mediators and glial activation in an Nrf2-dependent manner, while pharmacological inhibition of heme oxygenase 1 (HO-1) abolished these effects, highlighting the central role of the Nrf2/HO-1 axis [82]. Consistently, ICA reduced dopaminergic neurotoxicity and glial-driven neuroinflammation in a 6-hydroxydopamine-induced Parkinson’s disease model through Nrf2-dependent mechanisms [83], and similar anti-inflammatory and functional benefits were observed in models of spinal cord injury, where ICA treatment correlated with significant motor recovery [64,84]. Mechanistically, accumulating evidence indicates that ICA-mediated Nrf2 activation is regulated upstream by sirtuin 1 (SIRT1). In an amyotrophic lateral sclerosis model, inhibition of SIRT1 abolished ICA-induced Nrf2 activation and reversed motor improvements, demonstrating a functional dependence on the SIRT1/Nrf2 axis [85]. Supporting this hierarchical relationship, independent studies showed that pharmacological or genetic disruption of SIRT1 or Nrf2 signaling negated neuroprotective effects elicited by other compounds, confirming SIRT1 as an upstream regulator of Nrf2-mediated cytoprotective responses [86,87]. Notably, ICA does not alter Nrf2 mRNA expression but increases nuclear Nrf2 protein levels [82,83], indicating a post-transcriptional mechanism involving enhanced protein stability and nuclear translocation. Collectively, these findings support a model in which ICA attenuates glial-driven neuroinflammation and neurodegeneration by modulating the SIRT1/Nrf2/HO-1 axis, thereby restoring the balance between antioxidant and proinflammatory signaling pathways, including crosstalk with NF-κB, and highlighting this pathway as a promising therapeutic target in neuroinflammation-driven neurodegenerative diseases (Figure 2) [87].
In addition to the SIRT1/Nrf2/HO-1 axis, ICA also engages insulin-like growth factor 1 receptor (IGF-1R)–dependent signaling to limit neuroinflammation and excitotoxic damage. Icariin and icaritin suppressed lipopolysaccharide-induced expression of TNF-α, IL-1β, cyclooxygenase-2 (COX-2), and inducible nitric oxide synthase (iNOS), while enhancing basal phosphorylation of ERK1/2 and protein kinase B (AKT) [88]. Concomitantly, ICA upregulated the astrocytic glutamate transporters GLT-1 and GLAST, supporting improved glutamate clearance and neuronal protection. Importantly, all anti-inflammatory and regulatory effects were abolished by pharmacological IGF-1R inhibition, confirming IGF-1R–dependent signaling [88].
Taken together, these findings demonstrate that ICA targets neurodegeneration by converging on key inflammatory and oxidative pathways that link chronic glial activation to neuronal dysfunction. By suppressing proinflammatory signaling cascades such as NF-κB, cGAS–STING, and NLRP3 inflammasome activation, while simultaneously enhancing SIRT1/Nrf2/HO-1–mediated cytoprotective responses, ICA restores the balance between inflammatory and antioxidant signaling, preserving neuronal integrity and function in neurodegenerative conditions driven by sustained neuroinflammation [34,61,70,71,72,73,75,76,77,80,88,89]. Table 1 shows information from all the studies related to this topic.
3. ICA as a Potent Antioxidant Agent
Multiple animal and cellular models of nervous system disorders consistently demonstrate the disruption of redox signaling as a pathological hallmark. Called as oxidative stress, this is defined as a homeostatic imbalance between oxidizing molecules and antioxidant defenses, where the prevalence of ROS and NOS, such as hydrogen peroxide and peroxynitrite, results in molecular damage [93,94]. Oxidative stress is also frequently associated with an enzymatic imbalance, in which the excessive production of radicals by enzymes such as such as Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase and the dysfunction of the mitochondrial respiratory chain outweigh the neutralization capacity of endogenous antioxidant enzymes. These include Superoxide Dismutase (SOD), Catalase (CAT), and Glutathione Peroxidase (GSH-Px) [93,95,96]. As a signaling antioxidant response, cell signals activate specific intracellular pathways, mainly mediated by the translocation of the transcription factor Nrf2 to the nucleus [97]. There, by binding to the Antioxidant Response Element, Nrf2 induces the expression of protective genes (such as HO-1 and NQO1) [98], restoring cellular balance and preventing regulated cell death pathways.
Importantly, a growing body of evidence from cellular and animal models of neurodegenerative disorders demonstrates that ICA effectively counteracts oxidative stress-induced neuronal injury through multiple, partially overlapping signaling pathways. Across diverse experimental systems, ICA decreases ROS production [26,64,65,78,82,83,84,85,89,91,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116] and lipid peroxidation (malondialdehyde, MDA) [64,78,84,91,101,108,110,116]; preserves endogenous antioxidant defenses, including GSH [65,106,110,116], SOD [65,78,108,110,111,116], CAT [78,107], and GSH-Px [110,116]; and stabilizes mitochondrial function [65,84,108,111,115]. In addition, ICA modulates critical molecular nodes that connect oxidative stress to regulated forms of cell death, such as caspase-3 activation [26,78,84,101,106], p53 [106], glycogen synthase kinase-3 beta (GSK-3β)/Tubulin-associated unit (Tau) [111,113], sirtuins [65,107,116], and Nrf2 [82,83,105,116].
In vivo studies using senescence-accelerated mice (SAMP8) have shown that chronic ICA treatment enhances the activity of SOD and GSH-Px in brain tissue [111]. Similarly, in haloperidol-induced Parkinson’s models, ICA significantly increased the activity of glutathione S-transferase, CAT, and SOD in neuro-glial cells [108].It is important to note, however, that the efficacy of ICA may be context-dependent. For instance, in certain cerebral ischemia models, ICA monotherapy showed limited efficacy [104], whereas its combination with Panax notoginseng resulted in a synergistic reduction of oxidative stress markers [117]. This suggests that while ICA is a potent antioxidant modulator, its effectiveness can be influenced by the specific pathological environment or the presence of synergistic compounds.
Beyond systemic effects, in vitro studies suggest that ICA directly influences the antioxidant response of neurons. In PC12 cells treated with diabetic encephalopathy metabolites, ICA increased SOD activity and reduced MDA accumulation [109]. Similarly, in primary cortical neurons, ICA upregulated Prx1 mRNA and CAT activity via a SIRT1-dependent mechanism. Complementing these enzymatic responses, ICA prevents GSH depletion in PC12 cultures exposed to H2O2 by inhibiting the c-Jun N-terminal Kinase (JNK) and p38 Mitogen-Activated Protein Kinase (MAPK) pathways [107]. In spinal cord injury models, ICA’s antioxidant capacity was evidenced by its ability to prevent the reduction of GSH levels in neural tissue [64,84].
ICA also exerts neuroprotection by directly stabilizing mitochondrial function and preventing mitochondria-dependent apoptosis. In models of spinal cord injury and Sodium Azide (NaN3)-induced stress, ICA restored Mitochondrial Membrane Potential and enhanced ATP production [84,112]. ICA helps maintain mitochondrial homeostasis by inhibiting the formation of the mitochondrial permeability transition pore. Furthermore, ICA acts as an antagonist of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, specifically reducing excessive calcium influx [101]. By preventing this calcium overload, ICA averts the subsequent breakdown of Ca^2+^-dependent regulatory processes that typically triggers mitochondrial collapse. Corroborating this evidence, in APP/PS1 transgenic rats, ICA reduced intracellular iron accumulation, thereby limiting iron-catalyzed ROS generation (Fenton reaction) that typically damages mitochondrial membranes [115].
However, ICA can act as a sophisticated signaling modulator in the oxidative stress response. The most prominent signaling mechanism for ICA is the activation of the Nrf2 cascade. In different neurodegeneration models, ICA promoted Nrf2 nuclear translocation, leading to the expression of HO-1 and NQO1 [82,83,106]. This effect was notably absent in Nrf2-knockout models, where ICA’s protective effects were largely abolished. Another study related ICA action against neurotoxicity through the SIRT3/PGC-1α signaling axis [118], demonstrating SIRT-3 as an essential signaling axis for maintaining the GSH/GSSG ratio and reducing oxidative damage to mitochondrial membranes by PGC-1α. In rotenone-induced Parkinson’s models, ICA prevented the suppression of Sirtuin 3 (SIRT3) and Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha PGC-1α [65,82].
ICA activates the PI3K/Akt/GSK-3β survival pathway to protect neurons from NaN3)-induced apoptosis [112]. By inhibiting the c-Jun N-Terminal Kinase (JNK) and p38 MAPK pathways, ICA prevents DNA oxidation and subsequent cell death [107]. Reinforcing these findings, computational modeling reveals that ICA has a high binding affinity for targets associated with Brain-Derived Neurotrophic Factor (BDNF) signaling. This interaction likely enhances neuronal survival and plasticity, while simultaneously inhibiting Receptor for Advanced Glycation Endproducts (RAGE) and Glutamate Ionotropic Receptor AMPA Type Subunit 1 (GRIA1), both of which are central to redox deregulation [110,113].
Interestingly, ICA’s neuroprotective profile extends significantly to the glial microenvironment. In models of neuroinflammation, ICA administration reduced microglial populations and inhibited the release of pro-inflammatory mediators. In LPS-stimulated BV-2 microglia, ICA bolstered antioxidant capacity by upregulating the Nrf2/HO-1 pathway, effectively suppressing the “cytokine storm” associated with oxidative stress [92]. Crucially, in cuprizone-induced demyelination models, ICA treatment was associated with reduced ROS levels and enhanced maturation of oligodendrocyte progenitor cells. This suggests that ICA may promote a redox-permissive environment essential for oligodendrocyte metabolic health and subsequent remyelination [92]. By mitigating oxidative stress within the glial microenvironment, ICA likely provides an indirect, secondary layer of neuroprotection through the preservation of axonal insulation [26,85] (Figure 3).
These comparative findings reveal a conserved molecular logic underlying ICA’s efficacy across distinct pathologies. While both neurons and glia utilize the Nrf2 pathway, the outcomes differ while in glia, Nrf2 activation primarily dampens inflammation and supports remyelination [85,92], in neurons, it directly inhibits apoptosis and restores redox signaling. The comprehensive neuroprotective action of ICA, incorporating SIRT1 regulation, mitochondrial stabilization, and glial modulation, positions it as a robust candidate for treating diverse CNS neurodegenerative disorders [119,120] (Table 2).
4. ICA as a Negative Regulator of Apoptosis
Apoptosis is a tightly regulated form of programmed cell death essential for tissue homeostasis; however, its dysregulation is a central driver of neurodegenerative pathology. While classically categorized into intrinsic (mitochondrial) and extrinsic (death receptor) pathways, these signaling cascades do not operate in isolation but instead function as an integrated network [122,123]. For example, both pathways converge on the activation of executioner caspases, such as caspase-3, which orchestrate the final dismantling of the cell, including DNA fragmentation [122,123]. Cells possess an adaptive “brake” system mediated by Inhibitors of Apoptosis (IAPs), which counterbalance these cell death triggers. As reviewed by Marivin et al. (2012), IAPs [such as X-Linked Inhibitor of Apoptosis Protein (XIAP) and Cellular Inhibitor of Apoptosis Proteins 1 and 2 (cIAP1/2)] provide a critical adaptive response to oxidative stress by directly binding to and inhibiting processed caspases (3, 7, and 9) [124]. This mechanism allows neurons to tolerate transient stress without committing to cell death. Consequently, neuroprotective strategies must not only block upstream triggers like oxidative stress or ER stress but also reinforce this adaptive IAP-mediated threshold [125,126].
Apoptotic cell death in the pathogenesis of Alzheimer’s disease is associated with the activation of pro-apoptotic mediators, such as caspase-3 and -6, Bcl-2-associated X protein (Bax), and p53 upregulated modulator of apoptosis (PUMA), while anti-apoptotic mechanisms, such as B-cell lymphoma 2 (Bcl-2), Survivin and B-cell lymphoma-extra-large (Bcl-xL), are suppressed [127]. Several signaling pathways contribute to this imbalance, most notably PI3K/Akt, JNK/MAPK, and mechanistic target of rapamycin (mTOR). For instance, oxidative stress and inflammation drive activation of the JNK/MAPK pathway, which promotes apoptotic responses [127]. In parallel, impairment of the PI3K/Akt survival pathway enhances neuronal vulnerability by activating GSK-3β, thereby facilitating an Aβ/JNK/p53-induced cascade that culminates in neurofibrillary tangles (NFT) formation [127].
ICA demonstrates robust neuroprotective potential in Alzheimer’s disease models by mitigating core pathological hallmarks through the modulation of integrated apoptotic pathways. In vitro evidence in PC12 cell lines and primary cortical neurons indicates that ICA blocks amyloid-beta Aβ-induced neurotoxicity and sodium azide-induced mitochondrial dysfunction, resulting in the restoration of glucose metabolism and the reduction of tau protein hyperphosphorylation via activation of the PI3K/Akt/GSK3β pathway [106,112,114,128,129] (Table 3). These cellular findings are consistent with results from multiple in vivo models, including APP/PS1, 3xTg-AD, Tg2576, and SAMP8 mice. In these models, chronic ICA administration reduces APP expression and insoluble Aβ1-40 and Aβ1-42 levels [29,67,130,131] (Table 3). Specifically in the SAMP8 model, treatment attenuates memory impairment and reduces Aβ1-42 levels through the downregulation of BACE1, alongside increasing Bcl-2 expression and reducing Bax levels [67] (Table 3). Collectively, these actions promote the suppression of endoplasmic reticulum stress-induced apoptosis and the restoration of the Bax/Bcl-2 ratio, thereby preserving neuronal density in the hippocampus [29,130,131] (Table 3).
In the context of Parkinson’s disease, ICA consistently preserves dopaminergic integrity across various preclinical models by targeting pro-apoptotic factors that drive neuronal loss in the substantia nigra [25,132,134]. Studies utilizing 6-OHDA, Haloperidol, and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) models demonstrate that ICA administration, either alone or combined with levodopa, mitigates the loss of tyrosine hydroxylase-positive neurons and attenuates dopamine depletion in the striatum [25,132,134] (Table 3). These effects translate into the restoration of motor performance and enhanced antioxidant capacity, processes mediated by the activation of PI3K/Akt and MEK/ERK signaling pathways, as well as the inhibition of GSK3β activity [25,132,134] (Table 3). In PC12 cells, ICA confers cytoprotection against 6-OHDA-induced toxicity by reducing the Bax/Bcl-2 ratio and decreasing the proportion of cells in early apoptosis [25] (Table 3).
Advanced glycation end products (AGEs) are related to the occurrence of diabetic encephalopathy, a major complication of diabetes mellitus. Neuron apoptosis is a mechanistic factor on cognitive decline shown in streptozotocin induced animal models [143]. In a vitro model PC12 cell exposed to advanced glycation end products, ICA-treated groups had fewer apoptotic neurons and an overall reduction in caspase 3 and 9 levels. ICA directly inhibited Bax translocation to mitochondria, a mechanism central to its neuroprotection. Furthermore, ICA attenuated mitochondrial depolarization and restored antioxidant cell capacity, reinforcing its role in oxidative stress and apoptosis regulation [109]. Additionally, in primary hippocampal neuronal cell cultures from neonatal Sprague Dawley rats, Liu et al. (2011) reported that ICA suppressed corticosterone-induced apoptosis [134]. This protective effect was associated with inhibition of p38/MAPK activation and prevention of mitochondrial dysfunction, including preservation of mitochondrial membrane potential and suppression of caspase-3 activity [134]. In a similar model, it was demonstrated in neuronal primary hypothalamic cell culture that ICA prevented corticosterone-induced cell death via activation of the PI3-K/Akt pathway [135].
Cavernous nerve crush is an established model of axonotmesis [144], in which Wallerian degeneration and apoptosis of nitrergic nerves occur [145,146]. Conversely, in 12-week-old rats with cavernous nerve injury treated orally with ICA showed no differences in apoptosis markers in penile tissue homogenates [136]. However, the ICA-treated group exhibited a higher number of nerve fibers stained for neural nitric oxide synthase (nNOS) and upregulated nNOS expression, suggesting that ICA could rescue nitrergic neurons [136]. Moreover, in Wistar rats subjected to partial sciatic nerve ligation, chronic oral ICA administration reduced Bax and Bcl-2 protein levels and was associated with significant attenuation of neuropathic pain. This type of pain is suggested to activate the apoptotic pathway; thus, the reduction in these proteins could represent a greater protection of the sciatic nerve [73].
ICA demonstrated a robust neuroprotective effect against ischemic injury. Across diverse in vitro models, including oxygen–glucose deprivation/reperfusion (OGD/R) in neuronal cell lines (N2a [93] and PC12 [91,141]) and in primary cortical neurons [31,101,138]. In agreement, ICA benefits are also conserved in neonatal hypoxic–ischemic brain damage and adult middle cerebral artery occlusion in vivo models [138,139,140]. Treatment significantly improved cell viability by directly suppressing apoptotic pathways, as evidenced by dose-dependent reductions in cleaved caspase-3 and Bax expression, together with increased Bcl-2 levels [93,140,141]. Mechanistically activating pro-survival pathways while simultaneously suppressing proinflammatory and pro-apoptotic pathways (Table 3) [34,66,93,138]. In addition, ICA promotes cytoprotective autophagy through estrogen receptors (ERα/ERβ) activation and strengthens antioxidant defenses by upregulating Nrf2 and PPAR signaling [27]. ICA also exhibited a broader effects linked of endoplasmic reticulum stress pathways and pyruvate kinase M2 (PKM2)-dependent signaling [80,138], a conserved antioxidant action reducing ROS production [66,101,140,141], restored cytosolic Ca^2+^ homeostasis [101], and mitigated neuroinflammation [34,66,80,91,93,138] through inhibition of microglial activation, thereby preventing multiple apoptotic triggers [80,138]. These molecular changes translate into improved functional outcomes, including reduced infarct volume, diminished cerebral edema, and enhanced neurological performance.
Several apoptosis-related death models demonstrated that ICA can prevent neurodegeneration and also offer protection in non-degenerative neurological disorders such as depression [85], epilepsy [102] and schizophrenia [137]. ICA exhibits a robust multi-target neuroprotective profile mainly consistently targeting Bax/Bcl-2 axis and reducing caspase-3 activation across different pathological contexts (Figure 4). To mechanistically position ICA within the broader flavonoid signaling landscape, it is essential to synthesize its effects in contrast to the pro-apoptotic profile often reported in oncology. Recent comprehensive analyses describe a conserved pattern in neoplastic cells, where flavonoids such as ICA, quecetin [147], prunin [37] and isorhamnetin [119] induce apoptosis by inhibiting survival kinases, specifically the PI3K/Akt axis, suppressing NF-κB signaling and triggering mitochondrial dysfunction via reactive oxygen species accumulation. However, ICA exhibits a pathway-specific divergence in the CNS. Consistent with the neuroprotective signaling patterns reviewed for structurally related polyphenols, ICA sustains, rather than disrupts, PI3K/Akt and ERK1/2 phosphorylation in mature neuronal models [148]. This supports the hypothesis that flavonoid-driven modulation of redox and intrinsic apoptotic control is context-dependent: while these compounds exploit metabolic vulnerabilities to eliminate proliferating tumor cells, they reinforce intrinsic survival machinery in differentiated neurons. This duality highlights ICA as a promising agent for CNS preservation.
5. ICA and Autophagy
Autophagy is a lysosome-dependent degradation pathway essential for maintaining cellular homeostasis and can be classified into macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy depends on the Unc-51-like Autophagy Activating Kinase 1–Autophagy-Related Genes (ULK1-ATG) complex and recruitment of the class III PI3K complex for initiation and elongation of the autophagosome membrane [149]. Macroautophagy represents autophagy per se and is responsible for the elimination of deleterious proteins and dysfunctional organelles, such as mitochondria. The selective removal of damaged mitochondria, a process known as mitophagy, promotes mitochondrial quality control and involves upregulation of PINK1 and Parkin expression [150], leading to the targeted elimination of dysfunctional mitochondria before they can release pro-apoptotic factors [150]. In contrast, during microautophagy, the lysosomal membrane undergoes invagination or protrusion to directly engulf cytoplasmic contents [149]. Chaperone-mediated autophagy (CMA) targets proteins containing a Lysosomal Targeting Motif (KFERQ), recognized by Heat Shock Cognate Protein 70 (HSC70) and translocated into the lysosomal lumen via the Lysosomal-Associated Membrane Protein 2A (LAMP2A) [151].
The autophagic response is activated in the presence of cellular damage as an adaptive mechanism for cell survival. However, its role in cell survival is ambiguous and associated with autophagic flux. While basal autophagy acts as a cytoprotective mechanism for recycling damaged organelles/dysfunctional proteins [150], excessive or dysregulated autophagy can drive “autophagy-dependent cell death” [150]. In this context, therapeutic agents must have more than a binary influence on autophagic responses. Current evidence indicates that ICA functions as a context-dependent modulator, exerting opposing effects depending on whether the pathological state involves excessive activation of maladaptive autophagy or functional impairment of adaptive autophagy [152]. The outcome between survival and death is largely governed by the interaction between the autophagy initiator Beclin-1 and the anti-apoptotic protein Bcl-2 [153]. Under basal conditions, Bcl-2 binds to Beclin-1, preventing the assembly of the PI3K-III complex and inhibiting autophagy initiation. In conditions of acute stress, such as oxygen–glucose deprivation/reperfusion, this balance is disrupted [141].
Conversely, in chronic neurodegenerative models characterized by the accumulation of misfolded proteins, ICA appears to stimulate autophagy to facilitate adaptive clearance. For example, upregulating p62 in mice brain via gut microbiota manipulation mitigated amyloid-β toxicity and improved cognitive function in mice by enhancing autophagic clearance [154]. With regard to ICA, treatment has been shown to improve neuronal morphology, cell viability, and behavioral test performance in mice, likely by reducing amyloid-β accumulation in APP/PS1 transgenic models and Aβ1-42-treated PC12 cells. However, the upregulation of autophagy- and mitophagy-related proteins was observed only when ICA was combined with ꞵ-Asarone [155]. Notably, in a prenatal stress depression model, ICA treatment reduced hippocampal cell death and improved depressive-like behavior via modulation of Sirt-1/PGC-1α mitochondrial dynamics. Concurrently, ICA promoted mitophagy by increasing mitochondrial PINK1 and Parkin expression, elevating the Microtubule-Associated Protein 1 Light Chain 3, form II and I (LC3II/LC3I) ratio, and reducing P62 levels [156]. Interestingly, in Aβ1–42-injected rats, increased LC3 turnover was not followed by a decrease in P62, suggesting impaired autophagic flux in this Alzheimer’s disease model. In this context, ICA treatment reduced LC3-II, Cathepsin D, and Beclin-1 levels, while enhancing Akt and phosphorylated ribosomal protein S6 kinase bet-1 p-p70S6K activation, a key autophagy regulator, thereby reestablishing autophagic homeostasis [157].
ROS, largely produced in mitochondria, are well-established inducers of autophagy [158]. Keap1 acts as a redox sensor and induces Nrf2 degradation via proteasomal and autophagic pathways, thereby enabling its nuclear translocation. Nrf2 promotes the transcription of antioxidant response element (ARE)-driven genes, including p62, which functions as a selective autophagy adaptor by binding both ubiquitinated proteins and LC3, thereby accelerating autophagic clearance [159]. In addition, several Atg proteins are susceptible to cystine oxidation, indicating that ROS can directly modulate autophagic machinery [160]. ROS may also activate autophagy indirectly through inhibition of the PI3K/Akt/mTOR pathway or activation of MAPK signaling cascades, including JNK, p38, and ERK [160]. Oxygen–glucose deprivation/reperfusion (OGD/R) is characterized by increased ROS production and cell death [161]. PC12 cells exposed to OGD (2 h) followed by 24 h of reperfusion, increased apoptosis, and upregulation of the autophagy markers Beclin-1 and LC3-II were observed, suggesting an autophagy-dependent cell death. Pretreatment with ICA increased cell viability by upregulating Blc-2 and prevented excessive autophagy by reducing Beclin-1 and LC3-II levels. A similar effect was observed when 3-methyladenine (3-MA), an inhibitor of PI3K and autophagosome formation, was administered in the same model [162], indicating that ICA attenuates ROS-driven autophagy death [141]. In vivo evidence aligns with these findings. In a neonatal C57BL/6 model of hypoxic–ischemic brain damage (4 h induction), decreased Beclin-1 and LC3-II expression alongside increased p62 levels were observed, consistent with impaired autophagy [27]. ICA pretreatment ameliorated hypoxic–ischemic brain damage pathology by preventing these changes and concurrently upregulating the estrogen receptors ERα and ERβ. Importantly, administration of 3-MA abolished ICA’s protective effects, suggesting that ICA modulates macroautophagy in this context [27]. Notably, ICA’s effects were absent when animals were treated with mitochondrial processing peptidase (MPP) or PHTPP, selective antagonists of ERα and ERβ, respectively, indicating that ICA’s actions are at least partly mediated through estrogen receptor signaling [27] (Table 4).
Cellular senescence is driven by multiple mechanisms and exhibits diverse phenotypes, with autophagy initially proposed as a suppressor of senescence [170]. Experimentally, administration of D-galactose is widely used to induce accelerated cellular senescence in vitro and in vivo [171]. In an acute PC12 cell model, exposure to 200 mM D-galactose for 48 h significantly reduced cell viability and increased senescence markers such as p21 and senescence-associated β-galactosidase. These changes were accompanied by marked changes in autophagy-related proteins, including an increased LC3II/I ratio, decreased p62 levels, and the upregulation of Atg7, Atg5, and Beclin-1 proteins, suggesting an abnormal activation of autophagy flux. This dysregulation was likely a consequence of mitochondrial dysfunction, mitochondrial permeability transition pore (mPTP) opening, and ROS accumulation. Treatment with ICA effectively mitigated these effects by reducing ROS generation, autophagy marker expression, and cell death. Notably, the protective action of ICA seemed to be dependent on its ability to attenuate mPTP permeability and consequently promote autophagic homeostasis [172]. In vivo studies using the SAMP8 and ceramide-based senescence mice model further support ICA’s anti-senescent effects. Chronic ICA administration improved memory performance and reduced senescence markers such as SA-β-Gal and p21 in a p53-mediated process [173]. Moreover, ICA treatment reduced p62 and LC3-II protein expression, decreasing autophagosome formation and suggesting that ICA prevents senescence not by enhancing, but by modulating autophagic flux [35]. Collectively, these findings suggest that ICA may exert neuroprotective effects by fine-tuning autophagy to prevent maladaptive overactivation rather than by broadly increasing autophagic activity. More detailed information from studies related to this topic is found in Table 4.
6. Excitotoxicity and ICA
Excitotoxicity arises from an imbalance in excitatory–inhibitory neurotransmission, most commonly due to excessive glutamate, which leads to overstimulation of the postsynaptic neurons. This process triggers an abnormal influx of calcium ions and the activation of pro-apoptotic pathways involving p53, JNK/p38, and caspase-3 [107]. Exposure to high glutamate concentrations results in neuronal injury, a phenomenon strongly implicated in the pathogenesis of neurodegenerative diseases [174]. Recent evidence identifies ICA as a potential neuroprotective agent against excitotoxic cell death. In human SH-SY5Y neuroblastoma cells exposed to 10 mM glutamate, ICA treatment conferred dose-dependent protection by restoring antioxidant defenses; limiting intracellular calcium accumulation; upregulating phosphorylated ERK, CREB, and CaMKIIα; and reducing apoptosis-related protein expression [26]. Beyond glutamate-induced toxicity, ICA also demonstrated neuroprotection against other excitotoxic insults. In a methylmercury-induced model of amyotrophic lateral sclerosis, a motor neuron degenerative disease, ICA enhanced the expression of SIRT-1, Nrf-2, and HO-1 while decreasing TNF-α and IL-1β levels [85]. Similarly, in ibotenic acid-induced excitotoxicity, ICA increased the Bcl-2/Bax ratio and suppressed phosphorylation of Erk 1/2, JNK, and p38 proteins, all members of the MAPK family [163]. In another study using albino Wistar rats, ICA ameliorated ammonia–glutamate-induced excitotoxicity via its antioxidant properties and stimulation of the NO/cGMP pathway [164]. Collectively, these studies show how ICA can be a powerful agent against excitotoxicity and its death pathways.
7. ICA Regulates Endoplasmic Reticulum Stress Under Pathological Conditions
The endoplasmic reticulum is a dense, membrane-bound organelle that plays a pivotal role in calcium storage and in the synthesis of lipids and proteins, accounting for the production of more than one-third of all cellular proteins [175]. Within its lumen, newly synthesized proteins undergo proper folding and, frequently, post-translational modifications before being transported to their final destinations. Under pathological conditions such as oxidative stress, nutrient deprivation, or inflammation, the protein-folding capacity of the endoplasmic reticulum (ER) can become overwhelmed, leading to the accumulation of misfolded or unfolded proteins. This accumulation triggers a condition known as ER stress, which, in turn, activates the unfolded protein response (UPR), a signaling network able to restore ER homeostasis. The unfolded protein response enhances the ER’s folding capacity and reduces the burden of new protein synthesis through three primary sensors: PERK (protein kinase RNA-like endoplasmic reticulum kinase), IRE1α (inositol-requiring enzyme 1 alpha), and ATF6 (activating transcription factor 6) [176]. Under stress conditions, the dissociation of chaperones such as binding immunoglobulin protein (BiP)/GRP78 from these sensors enables their activation. IRE1α and PERK undergo trans-autophosphorylation, initiating signaling cascades that activate transcription factors including XBP1 and ATF4, respectively. PERK further attenuates global translation via phosphorylation of the initiation factor eIF2α, thereby reducing the load of emerging polypeptides entering the ER. In addition, IRE1α mediates regulated IRE1-dependent decay (RIDD), which degrades mRNA in order to reduce ER stress levels. ATF6, in turn, is transported to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases (S1P and S2P), releasing a soluble cytosolic fragment that functions as a transcription factor [177]. ER stress can also be counterbalanced by endoplasmic reticulum-associated degradation (ERAD) and reticulophagy. In the former case, misfolded or unfolded proteins are transported from the endoplasmic reticulum lumen to the cytosol through membrane-associated transporters, where they are ubiquitinated and subsequently degraded by proteasomes. In reticulophagy, segments of the endoplasmic reticulum containing unfolded or misfolded proteins are delivered to and fused with lysosomes, where they are subsequently degraded by acidic hydrolases [176,178]. However, if ER stress persists or becomes excessive, adaptive responses give way to apoptotic signaling. For example, these sensors share the ability to increase CHOP (C/EBP-homologous protein) expression. CHOP is a transcription factor that regulates the expression of pro-apoptotic proteins such as BAX, Bcl-2-interacting mediator of cell death (BIM), and PUMA, while suppressing anti-apoptotic proteins, including BCL-2 and BCL-XL, thereby promoting activation of the intrinsic apoptotic pathway [176,179]. Prolonged activation of IRE1α converts it into a signaling scaffold through its association with TNF-associated factor 2 (TRAF2) and ASK1, leading to JNK and p38 activation and the induction of mitochondria-dependent cell death via modulation of pro-apoptotic proteins such as BAX, BAK, BIM, and BH3-interacting domain death agonist (BID) [179]. The recruitment of TRAF2 by IRE1α also results in the dissociation of TRAF2 from caspase-12, which is localized at the ER membrane. Once released from TRAF2, caspase-12 undergoes facilitated activation, triggering the activation of caspase-9 and caspase-3 in a cytochrome c- and apoptotic protease activating factor 1 (Apaf-1)-independent manner. In addition, caspase-12 can be cleaved by calpains activated by calcium release from the endoplasmic reticulum lumen during ER stress [179]. Although prolonged ER stress is a classical inducer of apoptotic cell death, a growing body of evidence indicates that ER-induced cell death may also proceed through ferroptosis (see the Section 8). In this context, an imbalance in calcium and iron homeostasis induced by ER stress culminates in the production of ROS and the execution of ferroptosis [178].
Thus, pharmacological or molecular attenuation of ER stress represents a promising therapeutic strategy across a broad spectrum of diseases. Although relatively underexplored, current evidence indicates that the neuroprotective effects of ICA against neuronal loss are mediated, at least in part, through the inhibition of ER stress. ICA has been reported to attenuate ER stress in several experimental contexts, including models of Alzheimer’s disease [130], MCAO [80], SCI [33], OGD/R [138], and pharmacologically-induced ER stress [131,165]. Across these models, ICA treatment led to a marked reduction in the expression of key ER stress markers and associated apoptotic mediators, including GRP78, phosphorylated PERK (p-PERK), IRE1α, phosphorylated eIF2α (p-eIF2α), ATF4, XBP1, CHOP, and cleaved caspase-12 [130,131,165].
In rat hippocampal and cortical neuron cultures in which ER stress was triggered by corticotropin-releasing hormone (CRH) or OGD/R, ICA treatment effectively prevented ER stress progression by inhibiting NF-κB activation and, consequently, reducing the associated inflammatory response [138,166]. In PC12 cells, ICA administration for 24 h activated the transcription factor NFE2L1, leading to increased expression of synoviolin, an ER-anchored E3 ubiquitin ligase responsible for degrading misfolded proteins [131]. This ICA-induced upregulation of synoviolin conferred significant protection against ER stress-mediated apoptosis, an effect that was abolished when synoviolin expression was silenced via siRNA [131].
Another molecular target implicated in ICA’s regulation of ER stress is the PI3K/Akt signaling pathway. In a mouse model of SCI, ICA administration (50 µmol/kg/day) prevented injury-induced upregulation of IRE1α, ATF6, XBP1, eIF2α, GRP78, and CHOP. The ER stress induced by thapsigargin in cultured spinal cord neurons was also blocked by pretreatment with ICA in a PI3K/AKT pathway-dependent manner. The PI3K inhibitor LY294002 abolished ICA’s protective effect, indicating that the suppression of ER stress and the promotion of cell survival by ICA are dependent on PI3K/Akt signaling [33]. In addition to modulating intracellular signaling, ICA may directly interact with ER stress-related proteins. Molecular docking analyses demonstrated ICA’s ability to bind to the active sites of GRP78, IRE1α, and PERK, with calculated binding free energies of −8.87, −9.80, and −10.35 kcal/mol, respectively. ICA formed hydrogen bonds with Asp34 and Thr37 of GRP78; Glu651 and Cys645 of IRE1α; and Asp955, Cys891, and Met934 of PERK. These interactions suggest that ICA can directly regulate these key ER stress sensors, thereby suppressing ER stress-induced injury in models of ischemic stroke [80] (Figure 5). For more detailed information about studies discussed in the present topic, see Table 4.
Together, these findings indicate that ICA may exert its beneficial effects in CNS cells by preventing ER stress triggered by different insults, thereby advancing understanding of this flavonoid’s mechanism of action. However, further studies are needed to better elucidate the molecular mechanisms underlying ER stress inhibition.
8. Role of ICA in Ferroptosis
Ferroptosis is a non-apoptotic, regulated form of cell death that plays a pivotal role in degenerative diseases and malignancies [180]. It is an iron-dependent process characterized by extensive peroxidation of polyunsaturated fatty acids (PUFAs) within membrane phospholipids, leading to membrane destabilization and rupture independently of caspase activation [180,181]. The iron metabolism plays an important role in the execution of ferroptosis. Extracellular iron bound to transferrin is internalized via endocytosis through its interaction with Transferrin Receptor 1 (TfR1). Within endosomes, ferric iron (Fe^3+^) is reduced to ferrous iron (Fe^2+^) and exported to the cytosol by metal transport proteins (such as divalent metal transporter 1 (DMT1)) [180,182,183]. Subsequently, Fe^2+^ is sequestered within ferritin for storage, a process facilitated by interactions with poly(rC)-binding proteins [180]. This cytosolic Fe^2+^ is central to ferroptotic mechanisms, as it fuels the Fenton reaction, generating ROS such as peroxyl and hydroperoxyl radicals. These radicals catalyze the peroxidation of PUFAs, triggering a self-propagating chain reaction across the plasma membrane. Additionally, this process can be initiated by non-enzymatic Fenton-type reactions or enzymatically via lipoxygenases [180,184].
Another hallmark of ferroptosis is the reduction in antioxidant enzymes, particularly glutathione peroxidase 4 (GPX4), an enzyme that converts phospholipid peroxides into lipid alcohols, with expression controlled by selenium and GSH [185]. GSH is an essential cofactor for peroxidases, inhibiting lipid peroxidation and limiting the Fenton reaction by reducing hydroxyl radicals. GSH biosynthesis is regulated by cysteine metabolism, which involves cysteine acquisition via system X_c_^−^ or de novo synthesis through the trans-sulfuration pathway [180,186]. Once inside the cell, cysteine is processed by glutamate–cysteine ligase (GCL) and GSH synthetase (GSS) to generate the γ-glutamyl-cysteinyl-glycine tripeptide. In this way, ferroptosis inducers like erastin and sulfasalazine inhibit system X_c_^−^, preventing cystine import, while RAS-selective lethal 3 (RSL3) and statins induce ferroptosis via GPX4 inhibition [187,188].
Overexpression of ferritin heavy chain 1 (FTH1) in PC12 cells exposed to 6-OHDA, classically used to reproduce in vivo models of Parkinson’s disease, blocks ferroptosis and improves cell viability [189]. In a ferroptosis model induced by RSL3 in murine hippocampal HT22 cells, several flavonoids conferred protection. In contrast, ICA failed to prevent ferroptotic loss of cell viability, although only a single dose of 10 μM was tested, and higher concentrations may be required to prevent cell death [167]. Nevertheless, emerging evidence suggests a potential link between ICA and Mouse Double Minute 2 (MDM2)/Mouse Double Minute X (MDMX), negative regulators of p53 that promote ferroptotic death by altering membrane lipid composition. Inhibition of the MDM2/MDMX complex has been shown to lead to an accumulation of monounsaturated lipids, reduced CoQ_10_, and modify acylcarnitines and tri/diacylglycerols [190]. Network pharmacology analyses have identified MDM2 as a putative ICA target [36]. In APP/PS1 Alzheimer’s disease mice (10 months old), both ICA treatment and MDM2 knockdown prevented memory impairment, reduced intracellular iron accumulation, and restored antioxidant capacity [36]. Moreover, ICA and its glycosides effectively inhibit lipid peroxidation [191]. Specifically, ICA alone mitigates iron overload-induced mitochondrial damage through the modulation of ERK1/2/JNK-MAPK and PI3K/AKT/mTOR signaling pathways in bone marrow stromal cells [142,192]. Although the available evidence remains sparse, these findings collectively suggest that ICA may prevent ferroptotic cell death in neurodegenerative contexts, potentially through mechanisms involving iron homeostasis, antioxidant restoration, and MDM2 modulation. Further investigation into how ICA modulates ferroptosis-related markers—specifically ferritin, TfR1, GPX4, and the system x_c_^−^—is required to elucidate its therapeutic mechanism, as existing studies suggest its active role in regulating these pathways [168,169].
9. Conclusions
Traditional knowledge represents an invaluable resource for the identification of bioactive molecules with therapeutic potential, many of which are now globally available as dietary supplements, reflecting the widespread adoption of these practices. However, confirmation of these promising effects with scientific studies is important to ensure adequate, secure, and efficient use in patients across different disease contexts. ICA, a polyphenolic compound present in Epimedium-based formulations, has demonstrated broad systemic protective effects in a wide range of experimental models. In this review, we synthesized the molecular mechanisms through which ICA modulates neural cell death pathways. ICA attenuates neuroinflammation by reducing the expression of inflammatory mediators, including TNF-α, IL-1β, IL-6, iNOS, and COX-2, and by suppressing key signaling cascades such as NF-kB and TLR4. Interestingly, ICA also seemed to modulate the immune system systemically, by inhibiting Th1 and Th17 cell differentiation and suppressing inflammatory infiltration in the CNS. Therefore, ICA’s anti-inflammatory impact appears to be robust, modulating local and systemic responses. In this context, more studies are necessary to better describe and to confirm the contribution of systemic or local inflammatory modulations to ICA’s neuroprotective benefits in neurodegenerative diseases. In parallel, ICA counteracts oxidative stress by consistently reducing ROS, NO, and lipid peroxidation, while promoting Nrf2 nuclear translocation and the upregulation of antioxidant enzymes such as HO-1 and NQO1. This antioxidant response exhibits extensive crosstalk with inflammatory signaling and contributes to the modulation of excitotoxicity, ferroptosis, ER stress, and autophagy.
Preclinical evidence consistently indicates a robust neuroprotective effect of ICA against apoptosis in different models that mimic CNS pathologies. Its action is predominantly associated with the modulation of anti- and pro-apoptotic proteins, such as Bcl-2 and BAX, respectively. Additional evidence suggests that icariin inhibits apoptosis by reducing ER stress. However, other regulatory factors of the apoptotic pathway may also be modulated by ICA but have not yet been investigated. In this context, inhibitors of apoptosis proteins (IAPs), which suppress caspase activity, could potentially be upregulated by ICA. Another pro-apoptotic member of the Bcl-2 family that has gained increasing attention in recent years is Bcl-2-related ovarian killer (BOK). BOK is localized to the endoplasmic reticulum membrane and is capable of inducing ER stress-dependent apoptosis as well as mitochondrial outer membrane permeabilization independently of BAX and BAK. Therefore, evaluating the potential effects of ICA on BOK may provide novel and relevant insights into the mechanisms underlying the flavonoid’s modulation of cell death.
Another emerging topic in the literature is the inhibitory effect of ICA on ferroptosis-induced cell death. Evidence in the CNS remains limited and requires further investigation. There are currently no data demonstrating that ICA regulates components of system Xc^−^, a cystine transporter essential for glutathione synthesis, nor are there data regarding its effects on proteins involved in iron metabolism, such as ferritin and transferrin. Ferroptosis depends on polyunsaturated fatty acids in biological membranes, which are incorporated into these structures through specific enzymatic pathways. It is plausible that the inhibition of ferroptosis by ICA involves the regulation of these critical molecular targets required for the initiation of this form of cell death. Accordingly, investigating these pathways is essential to better understand the relationship between ICA and ferroptosis.
ICA further enhances cell survival by activating pro-survival signaling pathways, most notably PI3K/Akt/GSK-3β axis, with multiple studies indicating a strong dependence on SIRT-1 activation. As discussed here, computational modeling studies suggest that ICA can directly bind to proteins extremely important in neurodegenerative diseases, such as BDNF-related signals, RAGE, and glutamate ionotropic receptor AMPA type subunit 1. Therefore, besides the consistent antioxidant, anti-inflammatory, and anti-apoptotic signaling properties in the nervous system, ICA could promote neuroprotection by directly modulating key targets of neurodegeneration. However, the interpretation of these effects is complicated by the lack of standardization across experimental models, heterogeneous routes of administration, and wide variability in dosing regimens. Importantly, these limitations should be considered within a pharmacokinetic–pharmacodynamic framework. Low oral bioavailability, rapid systemic clearance, and limited neural tissue distribution impose constraints on ICA’s application as a conventional CNS-targeted therapeutic agent. The apparent requirement for higher doses in CNS-related models, compared with peripheral or ocular targets, likely reflects pharmacokinetic barriers rather than reduced intrinsic efficacy. In this context, extensive metabolism and the generation of bioactive derivatives, such as icariside I, icariside II, and icaritin, may substantially contribute to the neuroprotective effects attributed to ICA in vivo.
Despite the limitations of the preclinical evidence, the data are robust and support ICA as a promising candidate for neurodegenerative disorders. However, clinical research in neurodegenerative disease contexts remains largely unexplored. To date, only a single uncontrolled clinical study has reported a reduction in depressive symptoms in patients with bipolar disorder and comorbid alcohol use disorder following ICA administration [133]. Moreover, current clinical and translational studies rarely address the contribution of ICA metabolites or define outcome measures relevant to CNS disorders.
Collectively, current evidence indicates that ICA does not operate as a single-target neuroprotective agent but rather as a pleiotropic modulator whose efficacy is shaped by dose, tissue accessibility, metabolic conversion, and cellular context. Future investigations should therefore move beyond descriptive efficacy and prioritize direct comparisons between ICA and its major metabolites, dose-dependent and tissue-specific analyses, and pharmacokinetically informed experimental designs. Addressing these challenges will be essential to translate robust preclinical neuroprotection into clinically meaningful applications.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Vujosevic S. Limoli C. Kozak I. Hallmarks of Aging in Age-Related Macular Degeneration and Age-Related Neurological Disorders: Novel Insights into Common Mechanisms and Clinical Relevance EYE 20253984585910.1038/s 41433-024-03341-539289517 PMC 11933422 · doi ↗ · pubmed ↗
- 2Wang S. Jiang Y. Yang A. Meng F. Zhang J. The Expanding Burden of Neurodegenerative Diseases: An Unmet Medical and Social Need Aging Dis.2024162937295210.14336/AD.2024.107139571158 PMC 12339136 · doi ↗ · pubmed ↗
- 3Hacker K. The Burden of Chronic Disease Mayo Clin. Proc. Innov. Qual. Outcomes 2024811211910.1016/j.mayocpiqo.2023.08.00538304166 PMC 10830426 · doi ↗ · pubmed ↗
- 4Gorman A.M. Neuronal Cell Death in Neurodegenerative Diseases: Recurring Themes around Protein Handling J. Cell. Mol. Med.2008122263228010.1111/j.1582-4934.2008.00402.x 18624755 PMC 4514105 · doi ↗ · pubmed ↗
- 5Moujalled D. Strasser A. Liddell J.R. Molecular Mechanisms of Cell Death in Neurological Diseases Cell Death Differ.2021282029204410.1038/s 41418-021-00814-y 34099897 PMC 8257776 · doi ↗ · pubmed ↗
- 6Chi H. Chang H.-Y. Sang T.-K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases Int. J. Mol. Sci.201819308210.3390/ijms 1910308230304824 PMC 6213751 · doi ↗ · pubmed ↗
- 7Fricker M. Tolkovsky A.M. Borutaite V. Coleman M. Brown G.C. Neuronal Cell Death Physiol. Rev.20189881388010.1152/physrev.00011.201729488822 PMC 5966715 · doi ↗ · pubmed ↗
- 8Karvandi M.S. Sheikhzadeh Hesari F. Aref A.R. Mahdavi M. The Neuroprotective Effects of Targeting Key Factors of Neuronal Cell Death in Neurodegenerative Diseases: The Role of ER Stress, Oxidative Stress, and Neuroinflammation Front. Cell. Neurosci.202317110524710.3389/fncel.2023.110524736950516 PMC 10025411 · doi ↗ · pubmed ↗