GARD: Genomic Data-Based Drug Repurposing in Head and Neck Cancer with Large Language Model Validation
Pradham Tanikella, William Nenad, Christophe Courtine, Yifan Dai, Qingying Deng, Baiming Zou, Nosayaba Osazuwa-Peters, Travis P. Schrank, Di Wu

TL;DR
This study introduces GARD, a pipeline that uses genomic data and large language models to identify new drugs for head and neck cancer treatment.
Contribution
The novel GARD pipeline combines genomic analysis, network expansion, and LLM-based literature validation to accelerate drug repurposing for HNC.
Findings
GARD identified known and emerging drugs like Fostamatinib, Quercetin, and Aspirin for HNC treatment.
Genes like PIK3CA, SOX2, and TP53 were linked to HNC through genomic and network analysis.
HPV stratification improved the precision of drug repurposing in HNC subgroups.
Abstract
Head and neck cancer (HNC) is among the most prevalent and challenging cancers worldwide. Developing new drugs is expensive and time consuming, so this study explored a faster, cost-effective approach utilizing existing medications with established safety profiles: drug repurposing. We developed the GARDpipeline (Genomic Alteration-based Repurposing for Drugs), which utilizes large-scale genomic data from The Cancer Genome Atlas (TCGA) to identify key genomic changes in HNC. These genes are expanded through protein–protein interaction networks to capture related pathways and then validated using evidence from thousands of PubMed articles extracted by large language model (LLM) tools. Finally, validated genes are matched with drugs using the DrugBank database. This approach uncovered both known cancer drugs and promising new candidates. These included targeted therapies such as…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
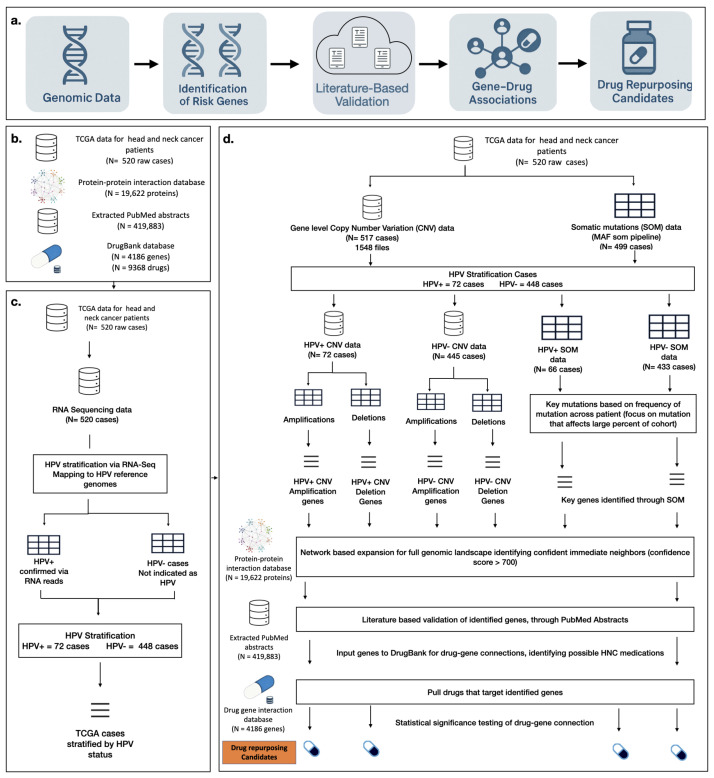 Figure 1
Figure 1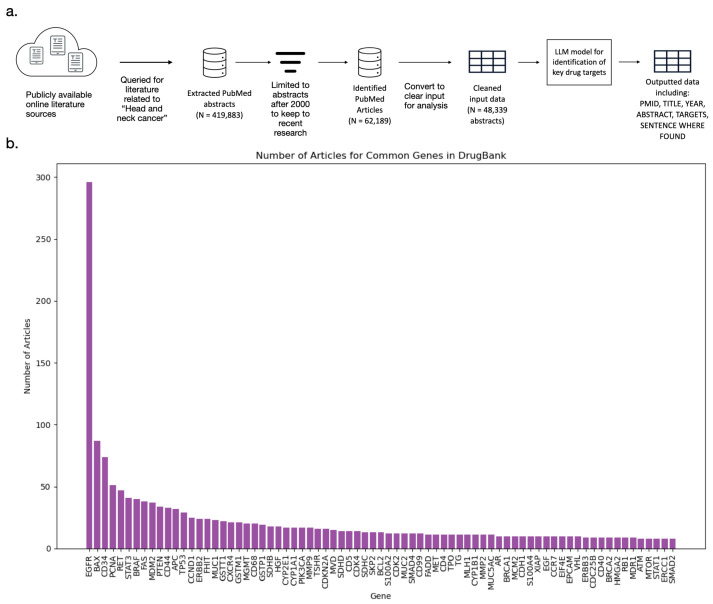 Figure 2
Figure 2 Figure 3
Figure 3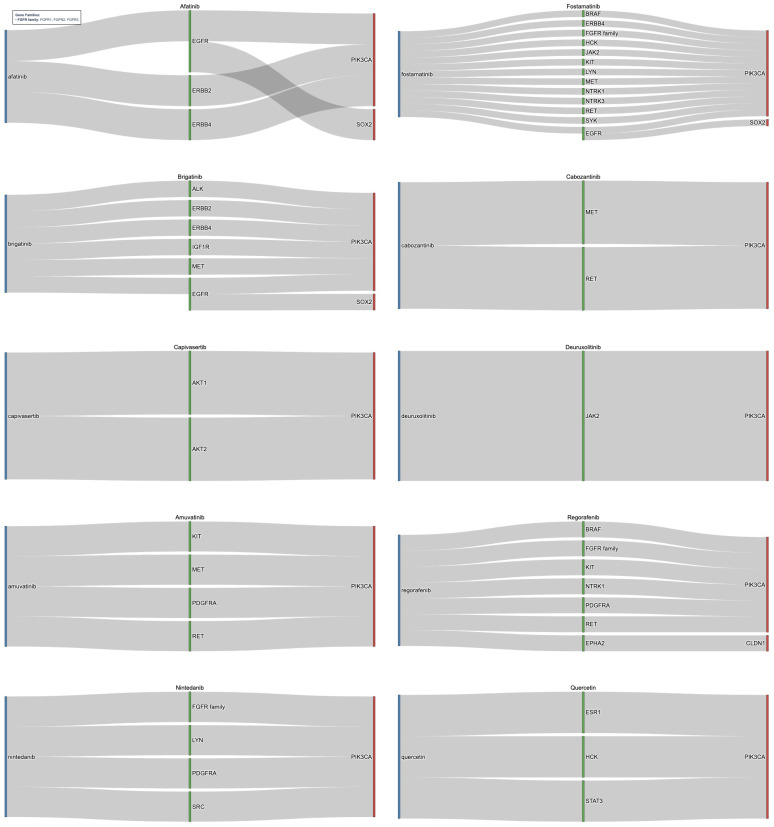 Figure 4
Figure 4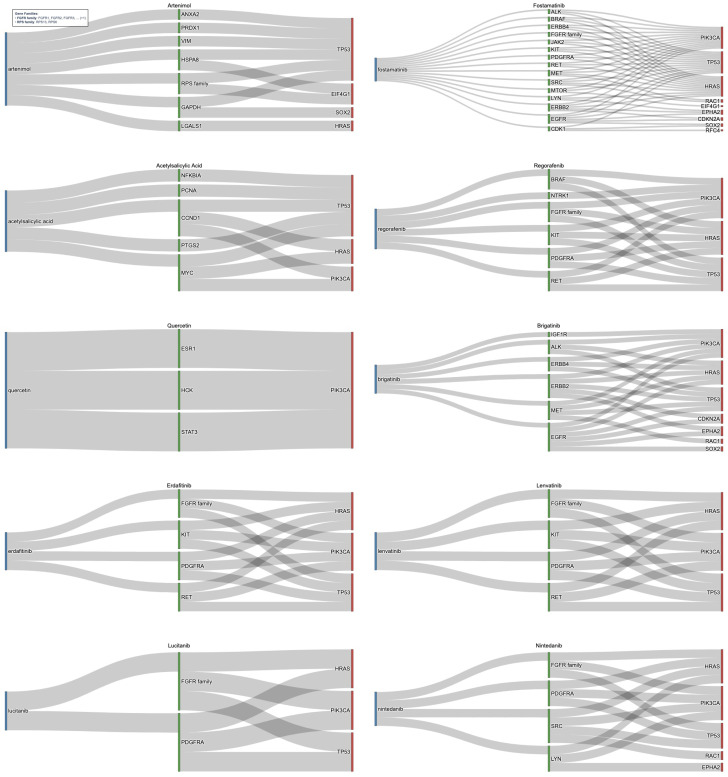 Figure 5
Figure 5- —University of North Carolina (UNC) Bioinformatics and Computational Biology T32-GM135123
- —UNC Lineberger Cancer Center pilot award
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHead and Neck Cancer Studies · Genomics and Rare Diseases · Cancer Genomics and Diagnostics
1. Introduction
Head and neck cancer (HNC) includes a diverse group of malignancies arising in the oral cavity, pharynx, hypopharynx, larynx, nasal cavity, and other regions of the head and neck, ranking as the seventh most common cancer globally [1]. Common treatments include surgical resection followed by radiotherapy and chemotherapy [2]. The medications/drugs in use today to treat HNC are still limited. Targeted therapies such as kinase inhibitors and immunotherapies have shown promise in other cancers and have been used in HNC recently [3]. Despite these advances, the treatment of HNC is still challenging due to the heterogeneity and novel treatments still have a hard time making it to market [2]. Additionally, a crucial factor to consider in HNC is HPV status, which significantly influences tumor biology, treatment response, and overall prognosis. Stratifying patients by HPV status enables more precise identification of genomic alterations and improves the relevance of therapeutic candidates for HPV-positive and HPV-negative subtypes [2,4].
Identifying more targeted treatments could significantly improve outcomes for patients with HNC. One promising and cost-effective strategy is drug repurposing, which involves finding new therapeutic uses for existing medications [5]. This strategy offers a cost-effective and time-efficient alternative to traditional drug development, as repurposed drugs have already undergone extensive safety and pharmacokinetic testing [6]. Computational repurposing approaches, which leverage large-scale biomedical datasets to systematically uncover mechanism-based drug–disease relationships, have shown potential [5,6,7,8]. Genomic-based computational methods include integration of genomic, transcriptomic, proteomic, and pharmacological data to identify drugs with potential efficacy against molecular targets. By harnessing publicly available multi-omics data, researchers can expand therapeutic options through the repositioning of approved or investigational compounds, ultimately advancing precision oncology [9,10].
In cancer research, genomic-based computational drug repurposing has already led to promising therapeutic advances. Recent advancements in available large-scale cancer genomics data have allowed for the investigation of cancers driving genetic therapeutic targets [11,12,13]. Cancer is driven by changes in the genome, and studying these changes in HNC can help identify key driver genes in tumor initiation and progression [2,14]. In this way, repurposed drug candidates that target key disease-related genes can be identified. The Cancer Genome Atlas (TCGA) and Cancer Cell Line Encyclopedia (CCLE) are some of the datasets that serve as sources for cancer-related genes. They provide information about copy number variations (CNVs), somatic mutations (SOMs), clinical data, and drug screens on cell lines [12,15,16].
The TCGA dataset serves as one of the main data sources for cancer-related genes. Existing TCGA-based drug repurposing varied in approaches and targets. Many studies focused on identifying therapeutic candidates in varied cancers, through differential gene expression towards patient outcomes such as binary cancer indicators, survival using linear models, or Cox regression models to identify genomic features associated with patient outcomes [17,18,19,20,21,22]. These and other studies focused around genomic-based analysis to identify underlying mechanisms and potential for use in treatment [20,23,24,25]. The use of CNVs or mutations from cancer genomics data for drug repurposing is less prevalent. Within HNC specifically, some studies narrowed in on specific medications or niche conditions [26]. Among these, one study maintained a broad approach to HNC alongside network expansion, as in this study, but did not incorporate HPV stratification and chose other methods of validation to land on very few specific repurposing candidates [19]. Some included HPV stratification, but were focused specifically on known or immediate alterations within the HNC genomic landscape and did not incorporate any network expansion [27].
Based on previous repurposing studies, a major limitation of current approaches lies in the lack of rigorous filtering and validation to prioritize a high-confidence set of drugs for clinical application [5]. Current works were able to identify possible repurposing candidates, but utilized one data source and needed larger manual review for identifying top candidates [5,15]. Additionally, many previous studies were effective in drug repurposing but had limited stratification of HPV analysis, or focused on survival-associated genes rather than a comprehensive genomic analysis, and only a few incorporated the use of robust validation steps to confirm candidate relevance and narrow repurposing candidates. The incorporation of network expansion to broadly identify possible HNC-targetable genes alongside HPV-stratified TCGA data with external validation provides a unique opportunity and approach to producing repurposing candidates. This study works to fill these gaps and presents a repurposing pipeline to produce a validated set of drug repurposing candidates from HPV-stratified TCGA genomics data.
This study introduces the GARD pipeline (Genomic Alteration-based Repurposing for Drugs) which integrates HPV stratification with large scale multi-omics to identify specific key disease related genes within HPV +/− patients. These genes are expanded using high confidence interactions with other proteins to ensure relevant biological pathways are included, and rigorously validated using thousands of peer-reviewed articles from PubMed. The use of PubMed literature in drug repurposing has had promising results, and the use of such methodologies as an exterior validatory method has the ability to ensure viable repurposing candidates are identified [28]. Lastly, the identification of medications is completed using DrugBank, where known drug–gene combinations are curated, and thorough empirical validation is performed. The results from our drug repurposing pipeline, GARD, confirmed the validity of its performance through the identification of both established and novel therapeutic candidates for HNC. In this study, we demonstrate that genomics-based computational drug repurposing can provide a scalable and precise framework for discovering new treatment options tailored to molecular subtypes, such as HPV-positive and HPV-negative HNC. These identified medications can serve as hypotheses of treatment avenues in HNC that warrant further exploration through clinical and experimental studies, with the goal of translating them into effective patient treatments. Given the heterogeneity of many cancer types and the growing availability of genomic datasets, the presented pipeline, GARD, holds promise for broader application across other malignancies and even non-cancer diseases, potentially accelerating the discovery of novel therapeutics, reducing development costs, and expanding precision medicine.
The GARD pipeline, including preprocessing scripts, statistical analysis modules, and visualization tools, is publicly available on GitHub at: https://github.com/pvtanike/Genomic-Landscape-Based-Drug-Repurposing.git (accessed on 17 February 2026).
2. Materials and Methods
2.1. GARD Pipeline Overview
To identify drug repurposing candidates for HNC, we developed the GARD pipeline. This framework integrates multiple publicly available biomedical resources to combine genomic insights with pharmacological and publicly available literature data for the identification of drug repurposing candidates in HNC.
First, at its core, GARD leverages TCGA, which provides multi-omics datasets, including Copy Number Variation (CNV) and Somatic Mutation (SOM) profiles with associated clinical annotations. The datasets were analyzed to produce high-confidence risk genes associated with HNC. All data was used alongside results of previous work to stratify patients by human papillomavirus (HPV) status [13]. This stratification enables subtype-specific analysis and improves therapeutic relevance [12].
Second, to capture pathway-level context, GARD expands high-confidence risk genes through STRING protein–protein interaction (PPI) networks, incorporating immediate neighbors with strong biological connectivity [29]. These genes are validated through external data sources of publicly available literature data within PubMed, as literature-based repurposing efforts have shown effectiveness [28,30].
Third, after extraction and cleaning, the literature data was parsed by the LLM model, Google GEMMA, to identify genes associated with HNC and provide peer-reviewed evidence as support for the gene association with HNC and filter the identified gene list for drug–gene associations [31].
Fourth, to connect these validated genetic findings with potential therapeutic agents, GARD incorporates curated drug–gene data from DrugBank [32]. To ensure that the drugs identified have a significant relationship with target genes identified either directly from CNV/mutation analysis or as the protein neighbor genes, statistical enrichment-based tests were performed for robustness.
Together, these resources form the foundation of a robust analytical framework designed to uncover clinically relevant drug repurposing opportunities and establish a scalable methodology applicable to other diseases. The approach centers on three key components: (1) extraction and preprocessing of genomic data, (2) analysis, identification, and validation of HNC-specific genotypic features, and (3) connecting with DrugBank to pinpoint viable drug repurposing opportunities. A clear diagram of the pipeline can be seen in Figure 1. A breakdown of the literature validation pipeline is available in Figure 2a.
2.2. TCGA Data
The key genomic data was sourced from TCGA, a comprehensive and collaborative initiative between the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI): https://www.cancer.gov/tcga (accessed on 13 November 2025) [12]. For this study, the TCGA HNC cohort corresponding to case IDs from the Nulton et al. study was utilized, allowing for HPV stratification. The final cohort included 520 cases, which were further refined based on the availability within each data category: copy number variation (CNV), and somatic mutation (SOM) data [12,13]. All data was accessed through the Genomic Data Commons (GDC) Data Portal. This study utilized open-access data, which includes de-identified clinical information, gene-level copy number variation (CNV) and somatic mutation profiles (SOM).
2.3. HPV Stratification
HPV serves as an influential factor in the diagnosis and treatment of HNC [1,2,33,34]. Its importance within the GARD pipeline was no different. HPV stratification was performed using results from Nulton et al., who demonstrated that RNA-Seq data can be used effectively to determine HPV status in HNC [13]. The majority of HPV-positive cases were HPV16, which was the primary genotype analyzed due to its prevalence and clinical relevance. Other HPV types were detected with low frequency. The Nulton et al. study analyzed 520 TCGA HNC cases and identified 72 HPV-positive patients [13].
2.4. Genomic Amplifications and Deletions
After stratification on HPV status, it was possible to utilize the data to identify key genomic components of HNC. Significant CNV amplifications and deletions were identified through statistical testing and GISTIC-like scoring, then used to highlight key genomic drivers and expanded to their high-confidence protein interaction neighbors for downstream therapeutic targeting. TCGA provides gene-level copy number variation (CNV) data, which quantifies changes in the number of copies of specific genes across tumor samples. Deviations from this baseline, either deletions (CNV < 2) or amplifications (CNV > 2), can indicate genomic alterations that are possibly associated with the cancer or disease being studied.
Based on the categorization from Nulton et al., the key genetic mutations were categorized into HPV+ and HPV- cohorts. Within the extracted HNC TCGA cohort, about 517 cases had cleaned CNV data. Within the available data for each sample, 58,645 available genes were analyzed and provided chromosome number, start and end position, and copy number value [12,35,36].
After identifying a gene as being amplified or deleted, it was important to determine whether the mutation was random or of significant prevalence across the entire disease landscape. In order to first filter to significantly altered regions for HNC, a significance test for high-level amplifications and deletions was calculated. High-level amplifications were considered as those with a CNV value greater than 4. Along the same lines, high-level deletions were considered as those equaling 0 [37]. To determine the significance of the mutation, the counts of high-level amplification or deletion were utilized to assess whether their occurrence across samples exceeded what would be expected by chance.
A binomial test was conducted where the significant mutation count per gene is the number of successes across all files. This approach enabled the derivation of a p-value to assess whether the CNV frequency in each gene exceeded what would be expected by chance, given the cohort’s overall genomic instability. The calculation of this value was:
where n is the total number of samples in the cohort, x is the number of samples with high-level CNV amplification (CNV copy number ) or deletion (CNV ) events, and p is the genome-wide background CNV rate calculated as for amplifications, and for deletions. This represented the proportion of all CNV measurements across all genes and samples that exceeded the high-level threshold. This approach tested each gene against the observed cohort-specific genomic instability.
Alongside this, an empirical null distribution was derived through permutation testing. For each 1000 permutations (selected to balance computational burden and significance), n CNV values were randomly sampled (with replacement) from the pooled set of CNV observations, and the count of sampled values exceeding the threshold (CNV > 4, CNV < 1) was recorded to build a null distribution. The empirical p-value was calculated by comparing the observed count to this null distribution. This was then divided by the number of permutations of the null distribution (n = 1000) to derive an empirical p-value. The empirical p-value was calculated as:
where is the observed number of samples with high-level CNV events for the gene, is the simulated count of high-level events in permutation i, is an indicator function that equals 1 when the simulated count equals or exceeds the observed count, and the “+1” in both numerator and denominator prevents p-values of exactly zero while maintaining proper probability bounds. The two p-value calculations were conducted across many genes, and each test had its own chance of producing false positives. As such, a false discovery rate (FDR) adjustment was necessary. The Benjamini–Hochberg correction was applied for this study [38].
To further filter key mutations, a methodology was developed based on the methodology within the GISTIC software (GISITC 2.0) [37]. The key aspect is that mutations are assigned a score that considers both amplitude and frequency. Amplification values are accumulated as log2 of the CNV value minus 1 to adjust for the diploid baseline. This helped to center the data around 2 or a diploid CNV count. A cap was placed on CNV values (CNV = 7) to ensure that outliers do not skew values and reduce visibility of genes with lower CNV amplitude but higher frequency. Deletion values were accumulated as +1 for minimal deletions (CNV = 1) and +2 for significant deletions (CNV = 0) to track the amplitude of CNV alteration. The frequency of significant mutations was calculated by tracking the number of times a gene was significantly amplified (CNV > 4) or deleted (CNV < 1). For amplification calculations, the score was calculated as the average log2-adjusted amplification value (amplitude) multiplied by the frequency percentage (prevalence) of high-level mutations. For deletions, the GISTIC-like score was calculated as the average absolute deletion intensity (amplitude) multiplied by the frequency percentage (prevalence) of significant deletion (CNV = 0) [37].
Significant genes were first filtered to those with significant mutations according to the FDR-adjusted p-values in both the binomial and empirical significance tests. To ensure robustness and control false discoveries, we implemented a conservative dual-testing approach requiring candidates to pass both parametric and non-parametric significance tests. To further refine candidate selection, the distributions of GISTIC-like scores for these genes were evaluated to assess the strength of copy number alterations. Rather than applying arbitrary fixed cutoffs, we employed a distribution-based approach where thresholds were determined empirically from observed data patterns. For each analysis stratum (HPV-positive/HPV-negative × amplification/deletion), we visualized score distributions using histograms, examined distribution statistics (mean, percentiles), and identified natural breaks where scores transitioned from the bulk distribution to the upper tail. The selected parameters proved ideal for identifying key risk genes. The final cutoffs were implemented to systematically identify top-priority genes within each subgroup. By integrating statistical significance with genomic alteration burden, this process prioritized a focused set of high-confidence genes most relevant to HNC. These key genes could serve as potential targets for medications and, in turn, identify possible repurposing candidates [5,39].
2.5. Somatic Mutations
In addition to the copy number variations (CNVs), somatic mutation (SOM) data was utilized. Somatic mutation (SOM) data was analyzed using length-normalized mutation burdens, statistical significance testing, and empirical permutation methods to identify key mutated genes in HNC, which were then expanded through high-confidence protein–protein interaction neighbors to support downstream therapeutic targeting. Somatic mutations were identified using cohort-level MAF analysis files from TCGA [13]. This identified cohort included information on a total of 16,477 mutations for the 499 cleaned cases available within the data. This data was stratified by the HPV status as described above, leading to 66 HPV-positive cases and 433 HPV-negative cases. After stratifying the data by HPV status, key genes were tracked by monitoring their mutations, using a method similar to the CNV tracking approach described in the above section.
The mutations were filtered to focus on non-synonymous mutation types, as synonymous mutations rarely impact protein function [40]. Gene lengths were extracted from GENCODE v48 annotations and used to normalize mutation counts, as longer genes have a greater opportunity to accumulate mutations by random chance simply due to larger mutational target size [41]. Each gene in the available data was analyzed to count the number of times a mutation occurs in a gene, as well as the unique number of patients harboring a mutation for the gene. Producing a mutation count and cohort frequency that were normalized by gene length.
A binomial test was then conducted to measure the enrichment of each mutation within the cohort and the significance of the mutation. Through the mutation-based analysis, the mutational burden of each gene within the cohort could be directly measured. This test observed whether the mutation count in a gene significantly exceeds the expected count based on its genomic target size. The test assumes the observed mutation count as the number of successes and the total number of mutations across the cohort as the trial size. The test statistic was calculated as:
where N is the total number of mutations observed in the cohort (including both synonymous and non-synonymous mutations to establish an unbiased background rate), x is the observed number of non-synonymous mutations in the gene, and p is the gene-specific probability calculated as the gene, where is the coding sequence (CDS) length of the gene, and the denominator represents the sum of CDS lengths across all protein-coding genes in the reference genome. This length-based analysis accounts for the fact that longer genes present larger mutational target spaces and are expected to accumulate more mutations by chance. The test directly measures whether the mutational burden in each gene exceeds expectations given its genomic target size.
As performed in the above section, an empirical significance test was conducted as well. For each gene, a multinomial null distribution is constructed based on the mutation cohort, the probability of gene mutation is based on gene length calculated as gene length/sum of gene lengths. The null was constructed for each gene to be the size of the number of simulations (n = 10,000). The number of simulations was increased to account for the sparse nature of mutation events in somatic data and balanced with computational capabilities. A multinomial distribution was used to account for different mutation probabilities based on gene lengths.
where is the total number of mutations observed across the cohort and is the gene-specific probabilities, with based on the CDS length of each gene. The multinomial distribution enables simultaneous simulation of mutation counts across all genes while preserving the constraint that total mutations remain constant and respecting differential mutational target sizes. The empirical p-value was calculated as:
where is the observed mutation count in the gene and is an indicator function that equals 1 when the simulated count equals or exceeds the observed count. As before, both binomial and empirical p-values were FDR corrected using the Benjamini–Hochberg procedure. Genes with FDR-adjusted p-values < 0.05 in both tests were retained as significantly mutated, ensuring stringent control over false discoveries while maintaining adequate statistical power [38]. These statistical tests were utilized to remove background noise and filter to significantly mutated genes in the studied cohorts. From here, candidates were identified using similar methodologies to CNV analysis. A composite mutation score was calculated for each gene as:
where Normalized Count represents the mutation burden per gene adjusted for coding sequence length ( ), and frequency percentage is the proportion of patients harboring mutations in the gene. This scoring framework balances mutation amplitude (burden per gene) with cohort prevalence (frequency across patients), ensuring that both recurrent and high-impact mutations are captured.
As with CNV analysis, genes were retained only if they passed both statistical tests, ensuring robustness. Following statistical filtering, top-priority genes were selected by applying cutoffs to the mutation score distribution, ensuring retention of genes with the highest mutational burden. To characterize mutation burden patterns, mutation score distributions were visualized and evaluated to identify natural inflection points marking separation between the bulk distribution and the upper-tail region enriched for biologically meaningful mutational events. The selected values provided balance between stringent filtering and biological coverage for identifying driver genes. These cutoffs were incorporated to systematically identify high-priority mutated genes within each analysis subgroup. Through integration of statistical significance with genomic alteration burden, this process prioritized a focused set of high-confidence genes that could be used to identify treatments for HNC and possible repurposing candidates.
2.6. STRING Protein–Protein Interaction (PPI) Network
Through the above methods, it was possible to identify key drug targets directly related to HNC. However, beyond the primary key genes directly implicated in HNC, their immediate network neighbors, genes connected either upstream or downstream in the relevant biological pathways, were also included in the analysis. This approach allowed for the consideration of both identified HNC direct risk genes and those with a single-degree interaction in the pathway context. Identifying these neighbors helps to account for other key genes that could potentially serve as drug targets [5,42]. Within the GARD pipeline, the STRING PPI network was utilized [29]. For this study, the STRING database was filtered to focus on homo sapiens (human) gene interactions and pathways (Version 12 accessed on 29 July 2025).
The direct risk genes identified through CNV and SOM mutations analysis were inputted to the STRING PPI to identify immediate neighbors of the genes. Once immediate neighbors were identified for the key genomic mutations, the top connections were limited based on their confidence score. Based on the STRING db documentation, a cutoff of 700, representing high-confidence connections while providing broader biological coverage than more restrictive thresholds, was used to keep top immediate neighbors [29]. The culminated top connected genes and key mutations of HNC were then utilized with the drug gene interaction database to identify medications that interact with these key targets.
2.7. LLM-Based Literature Extraction for Gene Validation
The literature-based validation pipeline was designed to complement genomic analyses by leveraging published research as an independent validation layer to verify and reinforce identified gene–HNC associations in drug candidates, ultimately identifying validated drug repurposing opportunities. The validation dataset was derived from publicly available resources in PubMed, a comprehensive repository including peer-reviewed biomedical and life sciences literature. The process begins with the large-scale acquisition of PubMed biomedical abstracts using NCBI’s E-utilities, a suite of tools designed for searching, retrieving, and analyzing PubMed records [30,43]. A query of “Head and Neck cancer” was provided to PubMed to extract all relevant literature for HNC key target identification (accessed on 6 August 2025). This query leveraged PubMed’s MeSH indexing system, which automatically maps search terms to controlled vocabulary, ensuring comprehensive capture of all relevant anatomic subtypes [44]. The query returned 419,883 abstracts, including titles, publication years, and abstracts. The dataset was then cleaned to remove incomplete or missing abstracts and filtered to include only studies published within the past 25 years (i.e., after the year 2000), ensuring relevance and contemporary scientific context. After cleaning and filtering, a total of 48,339 unique abstracts were retained for analysis.
The data was then structured for input to the model, which included essential metadata such as Pubmed ID (PMID), title, publication year, and abstract text, as shown in Table 1. After compiling and formatting the data that included abstracts related to HNC, the LLM Google GEMMA was utilized. The Google GEMMA 2B instruction version was utilized (gemma-2b-it). Google GEMMA has proven ability to interpret text and extract key information [31]. Due to the computational intensity of processing such a large dataset, the University of North Carolina at Chapel Hill (UNC) Longleaf High-Performance Computing Cluster was utilized to ensure efficiency and scalability.
The LLM was prompted to identify key therapeutic targets related to HNC and extract supporting evidence by locating the specific sentence within each abstract where the target was mentioned. The output structure, illustrated in Table 2, included the identified gene, the evidence sentence, PMID, and any associated drugs, enabling transparent verification and efficient downstream analysis. This output format was intentionally designed to streamline manual review by enabling the rapid inspection of gene–disease associations alongside their supporting sentences and source articles. This aided in ensuring appropriate validation occurred for associations identified through CNV and SOM analysis. The output structure facilitated this quick manual review. An overview of this design analysis can be seen in Figure 2a.
2.8. Drug–Gene Connection for Drugs in DrugBank
After identifying immediate neighbors through high-confidence STRING PPIs and validating both these neighbors and the top risk genes through literature-based analysis, the fully validated gene set was queried against DrugBank to determine drug–gene associations [29,32]. This ensured that both direct risk genes and their biologically relevant neighbors were incorporated, thereby broadening the landscape of potential repurposable drugs. We then assessed whether these validated genes corresponded to drug targets in DrugBank and evaluated the statistical significance of their drug–gene connections to identify therapeutically relevant agents and prioritize candidates for repurposing in HNC. DrugBank is a curated database, providing detailed annotations on experimentally validated drug–gene interactions, including the mechanism of action (e.g., inhibitor, activator, modulator). It contains information on 9368 medications and 4186 genes totaling 23,136 drug–gene interactions (Version 6.0 accessed on 31 July 2025).
To assess the statistical significance of drug–gene connections, two complementary enrichment approaches were applied. First, a right-tailed hypergeometric test via cumulative binomial distribution was used to evaluate whether a given drug targeted more risk genes than expected by chance, given all druggable genes in DrugBank as the background of the test. The method quantifies the likelihood of seeing the observed or more overlap in gene targets between the drug’s targets and risk genes, and to determine if this is more than expected by chance. Here, the null hypothesis stated that a drug’s target genes overlapped with the risk gene set randomly, while the alternative hypothesis indicated significant enrichment. The test statistic was calculated as:
where M is the total number of druggable genes in DrugBank (4186), K is the number of targets for a given drug, n is the number of risk genes (including PPI neighbors) present in DrugBank, and x is the observed overlap between the drug’s targets and the risk set. By normalizing for each drug’s total target count (K), this test inherently controls for medications with broad target profiles.
Second, an empirical permutation test was conducted to strengthen confidence in the results. In this test, random gene sets of equal size to the risk set were repeatedly sampled (100,000 permutations) from the background universe of drug targetable genes (all available genes within DrugBank). The large number of permutations was used here to ensure that the drug–gene associations were significant and ensure maximum precision with the computational power available. For each permutation, the overlap with drug targets was calculated. The empirical p-value was then defined as the proportion of permutations with an overlap greater than or equal to the observed overlap. This distribution-based approach controlled for potential biases in gene–drug network structure. The empirical p-value was then defined as:
where is the observed overlap and the indicator function equals 1 when the permuted overlap equals or exceeds the observed overlap. This empirical validation ensures that observed enrichment is not due to highly connected genes. This distribution-free approach provides robustness against deviations from hypergeometric assumptions and accounts for heterogeneity in drug target set sizes.
Both hypergeometric and empirical p-values were FDR corrected for using the Benjamini–Hochberg procedure. Drugs with FDR-adjusted p-values < 0.05 in both tests were retained as significant candidates [38]. This approach enabled the filtering of medications to identify those with significant drug–gene interactions. In conjunction with a set of genes identified through CNV and SOM analyses, as well as their immediate neighbors, these drugs demonstrate a strong association with HNC, with only those meeting both statistical enrichment and the literature validation of genes advancing as drug repurposing candidates. The medications hold therapeutic potential; those not originally indicated for HNC could serve as repurposing candidates.
3. Results
3.1. Overview of the GARD Pipeline
We developed the GARD pipeline to perform a comprehensive analysis of HNC using genomic data from TCGA, stratified by human papillomavirus (HPV) status. Through GARD’s multi-step framework, integrating copy number variation (CNV), somatic mutation (SOM) profiles, PPI networks, and curated drug–gene associations from DrugBank, the pipeline identifies both established and novel genetic targets with therapeutic potential. An overview of the pipeline can be seen in Figure 1.
Genomic alterations were categorized into three major types: copy number amplifications, copy number deletions, and somatic mutations. Each category was analyzed independently to identify statistically significant gene-level changes associated with disease progression. High-confidence associated genes were identified using a combination of binomial and empirical permutation testing, with FDR-adjusted p-values. This allowed for the removal of background noise and filtering for significantly mutated genes. The top significant genes were further filtered based on composite scores developed based on a combination of mutation amplitude (strength of mutation) and frequency (number of cases affected). These genes were then expanded through PPI networks in the STRING database using a PPI combined score > 700 as the cutoff for high-confidence interactions in PPI. This allows for the inclusion of immediate neighbors that may play biologically relevant roles in HNC pathogenesis [29].
After that, a key step within GARD of literature-based validation was performed for the above genes using PubMed abstracts parsed by the Google GEMMA LLM, which strengthens confidence in gene–disease associations and prioritizes biologically relevant targets [28,31]. Through large-scale analysis of PubMed abstracts, a comprehensive set of 6077 unique genes associated with HNC was identified. Figure 2b illustrates the top 75 genes, sorted by article frequency, highlighting the most extensively studied targets in HNC. The identified genes include well-established oncogenic drivers such as EGFR, TP53, PIK3CA, and CDKN2A, which are frequently implicated in tumor progression and therapeutic resistance. These findings confirm the robustness of the pipeline in capturing both key cancer genes and less-studied targets with potential biological significance. This literature-based validation step strengthens confidence in the genomic findings by providing independent evidence from peer-reviewed research.
Genes were then mapped to drug–gene associations using DrugBank, identifying both direct drug targets and network-derived candidates across HPV-positive and HPV-negative cohorts. To assess the statistical significance of drug–gene connections, two complementary enrichment approaches were applied: a right-tailed hypergeometric test to evaluate whether a given drug targeted more risk genes than expected by chance, and an empirical permutation test randomly sampling genes from the DrugBank universe compared observed overlaps with risk genes to that of the null distribution. DrugBank provided crucial data regarding drug–gene interactions and information on medications, including their toxicity, allowing for selection of ideal repurposing candidates. This integrative approach revealed distinct molecular vulnerabilities within each subgroup and prioritized medications with strong mechanistic relevance to the identified gene sets. The following sections incorporate these methods to identify results and repurposing candidates.
Table 3 summarizes the number of high-risk genes identified through CNV and somatic mutation analyses, their connected immediate neighbors, and the corresponding significant medications uncovered through enrichment-based filtering.
3.2. HPV-Positive Investigation
Analysis of HPV-positive samples using CNV and somatic mutation data identified distinct genomic alterations associated with head and neck cancer. Table 3 summarizes the cohort-level results, showing that the HPV-positive CNV amplification analysis yielded 169 high-risk genes, of which eight were available as drug targets, and led to 1874 immediate neighbors totaling 627 targetable genes, resulting in 143 significant medications. The CNV deletion analysis identified 274 high-risk genes, of which 20 were available as drug targets, and led to 1518 immediate neighbors totaling 458 targetable genes, resulting in 42 significant medications. Somatic mutation analysis revealed six high-risk genes, of which one was available as a drug target (PIK3CA) and led to 338 immediate protein neighbors totaling 135 targetable genes, resulting in 90 significant medications from DrugBank. Overall, directly targeting the identified risk genes and their protein neighbors led to a total of 168 drugs to be repurposed, which is still relatively large. Of these 168 drugs, 14 were identified by direct connection to HNC risk genes, and 163 were identified through indirect connection between disease and drugs via protein–protein interactions. There were nine drugs identified from both procedures: Buparlisib, CH-5132799, Cladribine, Copanlisib, Golotimod, NADH, TG-100801, Wortmannin, and XL765.
To further refine this candidate set, literature validation of the genes was applied. A total of five genes (Appendix A Table A1) were validated in the literature among the above 51 unique risk genes and found to be targetable or connected to targetable genes through PPI. Among the validated risk genes, somatic mutations identified PIK3CA; amplification analysis derived PIK3CA, RFC4, CLDN1 and SOX2; and deletions identified TLR7. From here, the 168 drugs were reduced to 90, of which 6 were found by directly targeting risk genes (shown in Appendix A Table A2) and 85 targeted the protein neighbors of identified risk genes through protein–protein interactions (shown in Appendix A Table A3). One medication was found to overlap between the two sets: XL765 (Voxtalisib).
Identified risk genes were utilized with DrugBank to identify medications that directly target risk genes (direct targets) and develop significant drug–gene associations. Drugs directly targeting risk genes are illustrated in Figure 3a, highlighting key genes with existing therapeutic agents and potential candidates for drug repurposing (Table in Appendix A Table A2). Among direct targets, PIK3CA was associated with medications such as Wortmannin, XL765 (Voxtalisib), Buparlisib, Copanlisib, and CH-5132799, with the latter three identified as inhibitors. Buparlisib and Copanlisib have progressed to initial trials in combination regimens [45,46,47,48], CH-5132799 has shown potent antitumor activity in preclinical models, though it remains investigational [49], and XL765 (Voxtalisib) shows promising PI3K inhibition [50,51]. Wortmannin, despite its PI3K inhibition, is limited by poor selectivity and toxicity [52]. In contrast, TLR7 was linked to Golotimod, an immune-modulatory agent that completed Phase II trials for oral mucositis in HNC patients [53].
Furthermore, identified risk genes were expanded through high-confidence PPI networks, incorporating biologically relevant neighbors (indirect targets). Through validation of immediate neighbors and the direct risk genes they were connected to, the 263 unique druggable immediate neighbors originally identified were reduced to 33. Figure 4 illustrates the relationships between drugs, their gene targets, and high-risk genes for the top candidates identified by the pipeline. Top candidates were selected from the top 50 medications ranked by FDR-adjusted significance of drug–gene associations in HPV-positive HNC (full list in Appendix A Table A3). The medications target critical oncogenic pathways stemming from connections to risk genes of PIK3CA, SOX2, and CLDN1 underscoring their role in HNC [54,55,56,57]. Notable candidates included kinase inhibitors such as Afatinib, Fostamatinib, Brigatinib, Cabozantinib, Capivasertib, Deuruxolitinib, Amuvatinib, Regorafenib, and Nintedanib that work to target critical nodes including EGFR, MET, RET, KIT, PDGFRA, IGF1R, ALK, JAK2/3, AKT iso-forms, and the ERBB, FGFR, NTRK families.
Afatinib, an EGFR/ERBB inhibitor, showed efficacy in recurrent/metastatic HNC in trials such as LUX-Head & Neck [58,59], while Cabozantinib improved progression-free survival when combined with cetuximab or immune checkpoint inhibitors [60,61,62]. Fostamatinib stands out for its broad multi-kinase inhibition and Phase II evaluation in solid tumors, including HNC [63]. Capivasertib, a potent AKT inhibitor, selectively targets AKT1 and AKT2 and has demonstrated preclinical efficacy in HNC models, highlighting its potential to disrupt PI3K/AKT-driven oncogenesis [64,65,66]. Deuruxolitinib, a JAK2 inhibitor, represents an emerging candidate due to its ability to modulate the JAK/STAT pathway, which is frequently activated in HPV-positive tumors and associated with immune evasion and inflammation, making it a rational choice for repurposing [3,67]. Nintedanib has progressed to Phase II clinical trials in HNC, owing to its FGFR-targeting activity, addressing a key vulnerability in HPV-positive disease [68]. Brigatinib, originally approved for non-small cell lung cancer, inhibits ALK and EGFR signaling, suggesting strong potential for overcoming resistance mechanisms in HPV-positive HNC [69,70]. Amuvatinib, a multi-targeted tyrosine kinase inhibitor, acts on KIT, PDGFRA, MET, and RET, positioning it as a promising candidate for repurposing [71]. Regorafenib interacts with KIT, PDGFRA, RET, EPHA2, and FGFR family members connected to PIK3CA and CLDN1, providing a mechanistic basis for its application in HNC [72]. Additionally, non-kinase agents like Quercetin, identified for its pro-apoptotic and antioxidant effects, found to target ESR1, HCK, and STAT3, linked to PIK3CA, shows potenital for HNC treatment beyond conventional kinase inhibition [68]. Within both the direct risk-gene targeting analysis and the protein interaction expanded pathway analysis, XL765 (Voxtalisib) consistently emerged as a top candidate, underscoring its potential as a promising therapeutic option [51].
When comparing these results to previous TCGA genomic investigation work by Sayans et al., the 10 relevant cancer risk genes they identified had overlap with these results on PIK3CA and SOX2 [25]. Due to the HPV stratification, GARD captures genomic alterations that are significant within each HPV specific biological context. As a result, key genes that emerge in an HPV-positive landscape may not appear significant in a non-stratified analysis, leading to differences between GARD’s results and previous non-stratified methodologies. This difference in methodology explains the minimal overlap with repurposing pipelines such as Wei et al. [19]. Though it maintained a similar pipeline structure, their work was based on transcriptomic overlap with drug-treated cell lines. Combined with the absence of HPV-based stratification in their analysis, it is therefore unsurprising that the resulting drug candidates showed no overlap with the HPV-positive candidates identified in our study. Overall, HPV-positive tumors identified key risk genes that were expanded to include key signaling and targetable pathways, supporting the identification of novel therapeutics. Many were known or in clinical trials, but those that were not held potential as repurposing candidates. These findings highlight the potential for rapid translation of repurposed drugs into clinical applications for HPV-positive HNC.
3.3. HPV-Negative Investigation
Similar to the HPV-positive analysis, CNV and SOM analysis identified the HNC-associated genes. Table 3 shows that HPV-negative CNV amplification analysis identified 446 high-risk genes, of which 25 were available as drug targets, and led to 2945 immediate neighbors, totaling 886 targetable genes, resulting in 90 significant medications. CNV deletion analysis yielded 318 high-risk genes, of which one was available as a drug target, and led to 460 immediate neighbors totaling 127 targetable genes, resulting in 30 significant medications. Somatic mutation analysis revealed 166 high-risk genes, of which 32 were available as drug targets, and led to 2253 immediate neighbors totaling 913 targetable genes, resulting in 42 significant medications. This led to a total of 110 medications to be repurposed that targeted identified risk genes or immediate protein neighbors. Of these 110 medications, 20 were identified by directly targeting risk genes, and 104 were identified by indirectly targeting protein neighbors of risk genes. An overlap was found across 14 medications, including: 3-isobutyl-1-methyl-7h-xanthine, Acetylsalicylic Acid, Biotin, Bisindolylmaleimide i, Caffeine, Dasatinib, Ethanol, Fludiazepam, Fostamatinib, Glutamic acid, NADH, Phenethyl Isothiocyanate, Regorafenib, and Wortmannin.
Through the literature analysis, this was further refined. A total of 20 genes (Appendix A Table A4) were validated among the 224 unique risk genes found to be targetable or connected to targetable genes through PPI. Among these genes, somatic mutations identified TP53, PDE3A, MAGEC1, EPHA2, COL1A2, HRAS, REG1A, LRP1B, RAC1, CDKN2A, and PIK3CA. Amplifications derived CLDN1, DVL3, EIF4G1, PDCD10, PRKCI, RFC4, SOX2, and PIK3CA (identified through two methods). Deletions were identified in CDKN2B and CDKN2A (identified through two methods). This allowed the 110 medications identified to be reduced to 62 medications, of which 7 were found through directly targeting risk genes (Appendix A Table A5) and 60 were found through indirect targeting of validated protein neighbors of key risk genes (Appendix A Table A6). There are five drugs identified from both procedures. These were Acetylsalicylic acid, Bisindolymalemide I, Dasatinib, Fostamatinib, and Regorafenib.
The medications directly targeting key risk genes (direct targets) were associated with the validated genes, EPHA2, PIK3CA, PRKCI, and TP53. Drugs directly targeting risk genes are shown in Figure 3b, with full details provided in Appendix A Table A5. The medications directly targeting risk genes included some overlap with HPV+ direct risk gene targeting candidates such as Wortmannin and XL765, but included further candidates such as Fostamatinib, Acetylsalicylic acid, Bisindolylmaleimide I, Dasatinib, and Regorafenib. Although Wortmannin shows a significant relationship to the risk genes, it remains limited by poor selectivity and toxicity [52]. XL765’s appearance in both cohorts shows a strong relationship to HNC-related genes such as PIK3CA and the potential for repurposing [50,73]. Fostamatinib remains a strong candidate, as it is broadly applicable to many genetic targets related to HNC, and is currently in trials for HNC [63]. Acetylsalicylic acid, commonly known as Aspirin, is a pain reducer with anti-inflammatory properties. With limited information regarding HNC, initial findings prove promising, and it has shown effectiveness in other types of cancers [74]. Bisindolylmaleimide I is a compound utilized for protein kinase inhibition. It is not currently used clinically, but warrants further investigation due to its interaction with PRKCI, but due to its lack of use as a medication, remains as a lower-priority repurposing candidate [75]. Dasatinib is a tyrosine kinase inhibitor used in the treatment of leukemia, and has reached phase II clinical trials for HNC [76]. Finally, Regorafenib appeared again and shows strong inhibition of EPHA2 [72].
Identified risk genes were expanded through PPI networks to incorporate relevant neighbors (indirect targets). Validation of immediate neighbors and the direct risk genes were connected to reduced the 472 unique druggable immediate neighbors originally identified to a focused 77. Figure 5, which highlights the top medications identified after network expansion, shows the relationship between drugs, their gene targets, and root high-risk genes. These medications were selected from the top 50 medications ranked by FDR-adjusted significance of drug–gene associations in HPV-negative HNC (full list in Appendix A Table A6). The top medications targeted pathways stemming from connections to risk genes of TP53, PIK3CA, EIF4G1, SOX2, HRAS, RFC4, RAC1, EPHA2, and CDKN2A.
Notable candidates included multi-kinase inhibitors such as Fostamatinib, Regorafenib, Brigatinib, Erdafitinib, Lucitanib, Lenvatinib, and Nintedanib, targeting validated neighbor genes like EGFR, SRC, KIT, RET, MET, HSPA8, STAT3, PDGFRA, and the FGFR, ERBB, and RPS families. Several of these agents, Fostamatnib, Regorafenib, Brigatinib, and Nintedanib, were common to both HPV-positive and HPV-negative cohorts, reinforcing their broad mechanistic relevance. Others, such as Lenvatinib and Erdafitinib, emerged as novel candidates for HPV-negative disease. Lenvatinib, targeting the FGFR family, PDGFRA KIT, and RET, has demonstrated efficacy in HNC as part of combination regimens in clinical trials [77,78]. Erdafitinib, found to target similar genes, shows potential for repurposing in solid tumors, including HNC [79]. Lucitanib was found to target the FGFR family and PDGFRA and has shown promise in clinical trials for HNC [80]. The candidates of Fostamatnib, Regorafenib, Brigatinib, and Nintedanib were found again between the HPV+ and HPV- cohorts and shown to interact with similar key genes, such as the FGFR family, KIT, RET, PDGFRA, and SRC [63,69,72,81,82]. Beyond kinase inhibitors, other significant medications were identified. Artenimol, originally developed as an antimalarial, exhibits anticancer activity through immune modulation, targeting genes connected to TP53 and EIF4G1 [83,84]. Quercetin, previously highlighted in HPV-positive analysis, reappeared as a candidate due to its role in PI3K/AKT and STAT3 signaling [68]. Finally, Acetylsalicylic Acid (Aspirin) was found to target pathways connected to TP53, HRAS, and PIK3CA, reinforcing its potential anticancer activity and candidacy for repurposing in HNC [74]. Candidates that appeared in both avenues of analysis (direct and indirect gene targeting) included Acetylsalicylic acid, Bisindolymalemide I, Dasatinib, Fostamatinib, and Regorafenib. Many of these were strong candidates to be taken forward for analysis or are already in trials or investigations for HNC [63,72,74].
When compared with the results from the TCGA genomic study performed by Sayans et al., among their 10 key resulting genes, an overlap of TP53, CDKN2A, CDKN2B, PIK3CA, and SOX2 was observed [25]. This highlighted the ability of the pipeline to identify key genomic alterations within TCGA data. Studies, such as that published by Wei et al., show a TCGA-genomic-based cancer repurposing pipeline for HNC [19]. Though it was based around a similar pipeline, including a step for PPI expansion, this work was based on transcriptomic similarity between HNC patients in TCGA and the drug-treated cell lines. Therefore, it is not surprising that we find only one of the 22 repurposed candidate drugs for HNC identified in our drug candidates. Resveratrol, though not among the top candidates, was present among the top 50 HPV-negative indirectly repurposed drug candidates. Through the identified risk genes, alongside PPI-expanded gene pathways, key therapeutic targets were identified within the HPV-negative tumors. This led to the identification of known, or under investigation, medications alongside possible novel or exploratory repurposing candidates.
4. Discussion
With the expansion of available databases to utilize, drug repurposing has become a more popular and feasible way of discovering novel treatments for cancers and diseases [5]. This study introduces a genomics-driven, network-informed pipeline, GARD (Genomic Alteration-based Repurposing for Drugs), designed to systematically identify repurposing candidates for head and neck cancer (HNC). By integrating multi-omics data from HPV-stratified TCGA cohorts with high-confidence PPI networks and literature-based validation, GARD captures subtype-specific molecular signatures and expands them into biologically relevant networks. This multi-layered approach enables the identification of both direct genomic drivers and compensatory signaling hubs, which are then mapped to curated drug–gene associations from DrugBank, providing a robust framework for precision drug repurposing in HNC.
The analysis of the HPV-positive HNC cohort revealed a distinct molecular profile characterized by PIK3CA, SOX2, RFC4, CLDN1, and TLR7. Many were known cancer- or HNC-associated genes, but the incorporation of genes such as TLR7 presented underexplored targeting avenues [56,57,73,85,86,87,88]. These genes were expanded through PPI networks to further identify biologically relevant neighbors, and potential new therapeutic targets. These expanded genes included critical signaling mediators and pathway regulators (e.g., EGFR, KIT, JAK2/3, MET, RET, EPHA2, FGFR family, PDGFRA, and STAT3), which play essential roles in oncogenesis and therapeutic resistance [27,82,89,90,91]. Literature validation reinforced the mechanistic relevance of these targets, highlighting opportunities for precision drug repurposing in HPV-positive HNC. These results highlighted known pathways while uncovering novel targets, creating opportunities to identify repurposing candidates by either leveraging newly discovered gene targets or repositioning medications that act on established targets. The pipeline demonstrated its effectiveness by identifying drugs already in clinical trials for HNC, such as Buparlisib, Copanlisisb, Afatinib, Cabozantinib, Nintedanib, and Fostamatinib [45,48,59,62,81]. At the same time, it revealed promising new candidates, including XL765(Voxtalisib), Brigatinib, Amuvatinib, Deuruxolitinib, Capivasertib, and Regorafenib, which interact with critical signaling networks and represent opportunities for innovative repurposing strategies [50,67,69,71,72]. Beyond kinase inhibitors, Golotimod emerged as a unique candidate for immune modulation through TLR7 targeting [86,92]. Quercetin was also identified as a non-kinase agent with antioxidant and pro-apoptotic properties, offering unique potential to modulate PI3K/AKT and inflammatory pathways [68].
In HPV-negative tumors, alterations were dominated by cell-cycle regulators such as TP53 and CDKN2A/B, along with oncogenic drivers including PIK3CA, PRKCI, stem cell regulators like SOX2, tight-junction and EMT regulators such as CLDN1, and receptor tyrosine kinase nodes such as EPHA2 [14,56,57,73,89,93,94]. These genes were expanded through high-confidence PPI networks, revealing additional biologically relevant neighbors involved in critical pathways such as ERBB2/4, RET, KIT, EGFR, PDGFRA, MET, SRC, STAT3, FGFR family, and RPS family [27,82,90,91,95]. This network expansion broadened the therapeutic landscape by incorporating both known drivers and novel targeting opportunities not traditionally associated with HNC, such as EIF4G1. Literature-based validation reinforced the mechanistic relevance of these targets, highlighting opportunities for precision drug repurposing in HPV-negative disease. The pipeline confirmed its predictive strength by identifying drugs already in clinical trials for HNC, including Fostamatinib, Dasatinib, Lucitanib, and Lenvatinib [63,76,77,78,80]. At the same time, it uncovered novel candidates such as Brigatinib, Regorafenib, Erdafitinib, Amuvatinib, Artenimol, Acetylsalicylic acid, and Quercetin, which expand therapeutic options through possible repurposing candidates [50,67,69,71,72]. Notably, Erdafitinib, an FGFR inhibitor approved for bladder cancer, and Brigatinib, an ALK/EGFR inhibitor used in lung cancer, represent strong mechanistic candidates for repurposing in HPV-negative HNC. Beyond kinase inhibitors, Quercetin emerged as an agent with antioxidant and pro-apoptotic properties, offering unique potential to modulate PI3K/AKT and inflammatory pathways [68]. Similarly, Artenimol, originally developed as an antimalarial, demonstrated anticancer activity through oxidative stress and immune modulation, while Acetylsalicylic Acid (Aspirin) offers a low-cost anti-inflammatory strategy with emerging evidence of anticancer benefits [74,83,84].
By integrating direct genomic targets with network-based expansion, the GARD pipeline broadens the identified genes beyond known drivers, incorporating biologically relevant neighbors. The inclusion of literature-based validation further strengthens confidence in these identified targets, reducing false positives and prioritizing high-confidence targets. Identifying these gene targets and protein neighbors associated with HNC provides insight into the disease, enabling identification of drugs that can modulate these pathways, providing evidence for making them strong candidates for repurposing. This multi-layered approach allowed for the identification of known medications alongside novel candidates that have clear safety profiles and mechanistic pathways, improving the potential for translation into clinical practice. Importantly, the stratification of cohorts by HPV status proved essential for revealing distinct molecular vulnerabilities. HPV-positive and HPV-negative tumors exhibit fundamentally different drivers, which influence treatment response. By capturing these subtype-specific signatures, the GARD pipeline enables precision drug repurposing tailored to the unique biology of each subgroup, moving beyond one-size-fits-all approaches. This positions the pipeline as an advancement over previous genomic-based repurposing pipelines by filling the gaps of thorough genomic analysis of HPV stratified cohorts, inclusion of expanded relevant biological pathways, and comprehensive validation efforts. Overall, the integration of genomics data, network-based expansion, and literature validation through the GARD pipeline provides a robust and scalable framework for systematic drug repurposing in HNC and broader application across other malignancies.
This study’s reliance on TCGA data introduces limitations related to cohort size and population diversity, as such expansion through new databases or larger cohorts could provide expanded results. The reliance on HPV stratification through RNA-seq alignment carries the limitation of potential false identifications. The literature-based validation methods hold limitations to identify associations between genes and HNC, requiring manual review. The use of emerging LLM models would aid in improving the validation of causal relationships. These limitations lead the identified repurposing candidates to require further investigation. Currently, the repurposing pipeline is focused on one publicly available genomic dataset for identifying candidates and could benefit from expansion through transcriptomic or proteomic data, and other computational methods such as through real-world and EHR data [5,96]. Furthermore, while the computational predictions presented here are promising, experimental validation remains essential. Future work should focus on further validating data sources and translating these predictions into preclinical models, such as through HPV-stratified cell lines and adaptive clinical trial designs, to confirm efficacy and their use in clinical practice as viable therapeutic options. The identified candidates were a focused set of drug repurposing candidates that serve as a hypothesis to be taken through further computational, experimental and clinical evaluation. Through these methods, the identified candidates can be evaluated and validated further to translate the medication to clinical application in HNC.
5. Conclusions
In conclusion, the GARD pipeline identified 90 candidate medications for HPV+ HNC and 62 medications for HPV- HNC. Many medications were in initial trials for HNC and showed the viability of the pipeline for the identification of repurposing candidates. Further underexplored medications have the potential to serve as repurposing candidates in the field of HNC. The GARD pipeline demonstrates the potential for genomic-based drug repurposing and shows its potential in identifying novel treatments in HNC or further diseases.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Barsouk A. Aluru J.S. Rawla P. Saginala K. Barsouk A. Epidemiology, Risk Factors, and Prevention of Head and Neck Squamous Cell Carcinoma Med. Sci.2023114210.3390/medsci 11020042 PMC 1030413737367741 · doi ↗ · pubmed ↗
- 2Mody M.D. Rocco J.W. Yom S.S. Haddad R.I. Saba N.F. Head and neck cancer Lancet 20213982289229910.1016/S 0140-6736(21)01550-634562395 · doi ↗ · pubmed ↗
- 3Li Q. Tie Y. Alu A. Ma X. Shi H. Targeted therapy for head and neck cancer: Signaling pathways and clinical studies Signal Transduct. Target. Ther.202383110.1038/s 41392-022-01297-036646686 PMC 9842704 · doi ↗ · pubmed ↗
- 4Ndiaye C. Mena M. Alemany L. Arbyn M. CastellsaguéX. Laporte L. Bosch F.X. de SanjoséS. Trottier H. HPV DNA, E 6/E 7 m RNA, and p 16INK 4a detection in head and neck cancers: A systematic review and meta-analysis Lancet Oncol.2014151319133110.1016/S 1470-2045(14)70471-125439690 · doi ↗ · pubmed ↗
- 5Pushpakom S. Iorio F. Eyers P.A. Escott K.J. Hopper S. Wells A. Doig A. Guilliams T. Latimer J. Mc Namee C. Drug repurposing: Progress, challenges and recommendations Nat. Rev. Drug Discov.201918415810.1038/nrd.2018.16830310233 · doi ↗ · pubmed ↗
- 6Cha Y. Erez T. Reynolds I.J. Kumar D. Ross J. Koytiger G. Kusko R. Zeskind B. Risso S. Kagan E. Drug repurposing from the perspective of pharmaceutical companies Br J Pharmacol 201817516818010.1111/bph.1379828369768 PMC 5758385 · doi ↗ · pubmed ↗
- 7Brazil R. Repurposing Viagra: The ‘little blue pill’ for all ills?Drug Discov. Dev.2018 Available online: https://pharmaceutical-journal.com/article/feature/repurposing-viagra-the-little-blue-pill-for-all-ills(accessed on 17 February 2026)
- 8Rivero-Garcia I. Castresana-Aguirre M. Guglielmo L. Guala D. Sonnhammer E.L.L. Drug repurposing improves disease targeting 11-fold and can be augmented by network module targeting, applied to COVID-19Sci. Rep.2021112068710.1038/s 41598-021-99721-y 34667255 PMC 8526804 · doi ↗ · pubmed ↗