Gut Microbiome Signatures Across Migratory, Sedentary, and Aquaculture Ecotypes of Coilia nasus
Xue Liu, Congping Ying, Fengjiao Ma, Yanping Yang, Kai Liu

TL;DR
This study explores how the gut microbiome of Coilia nasus varies across different ecological types, revealing insights into their adaptation and conservation.
Contribution
The study identifies gut microbiome signatures linked to different ecotypes of Coilia nasus, offering new molecular markers for ecological tracing and conservation.
Findings
The core gut microbiota of Coilia nasus includes Firmicutes, Proteobacteria, and Actinobacteria.
Marine-adapted bacteria like Proteobacteria and Psychrobacter are enriched in migratory marine populations due to high salinity.
Aquaculture-reared fish show higher Actinobacteria abundance, likely due to artificial feed.
Abstract
This study constructed a database of intestinal microbiota for three ecological types of Coilia nasus, namely migratory type (comprising marine population and freshwater population), sedentary type and aquaculture-reared type, through 16S rRNA amplicon sequencing technology. This study investigates the ecological mechanisms underlying microbiota differentiation, focusing on three key drivers: environmental selection, host nutritional metabolism requirements, and host life history strategies. The results showed that the core flora of Coilia nasus consisted of Firmicutes, Proteobacteria, and Actinobacteria. Both the depletion of microbial taxa and the enrichment of marine-adapted bacterial lineages—including Proteobacteria and Psychrobacter—may be associated with elevated salinity in the migratory marine population of Coilia nasus. Significant variations in both richness and diversity of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1 Figure 2
Figure 2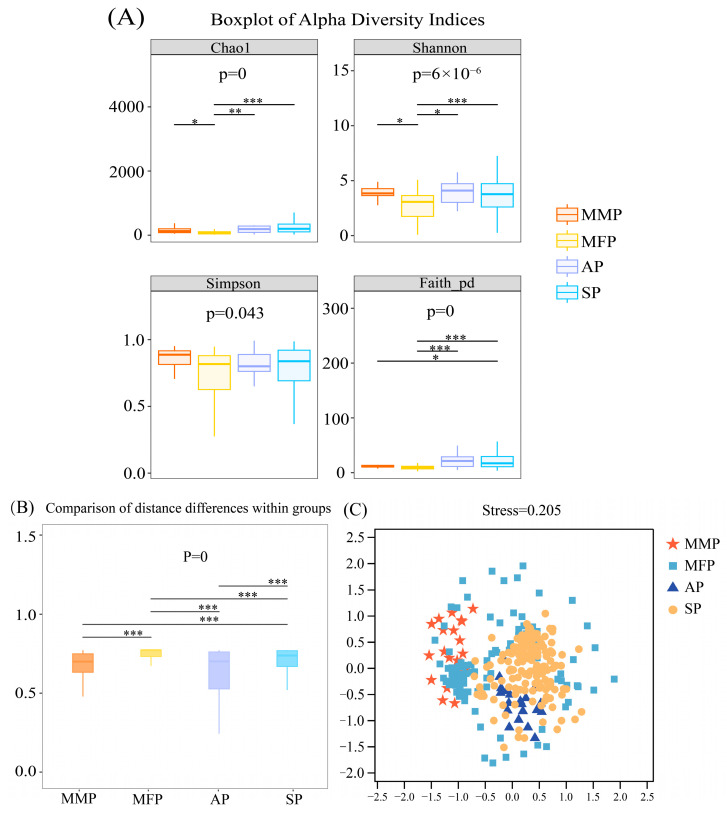 Figure 3
Figure 3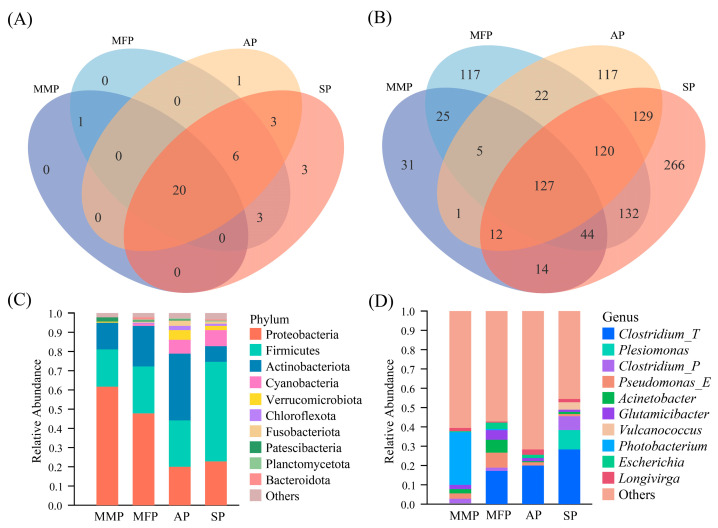 Figure 4
Figure 4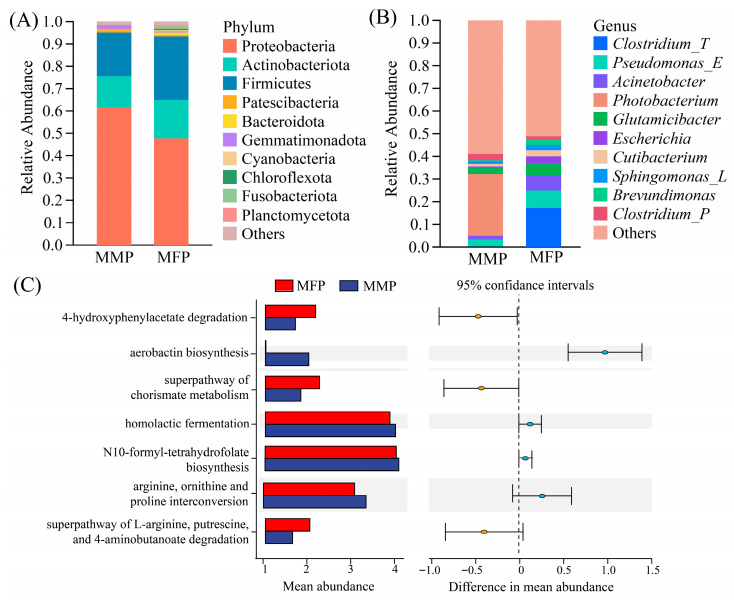 Figure 5
Figure 5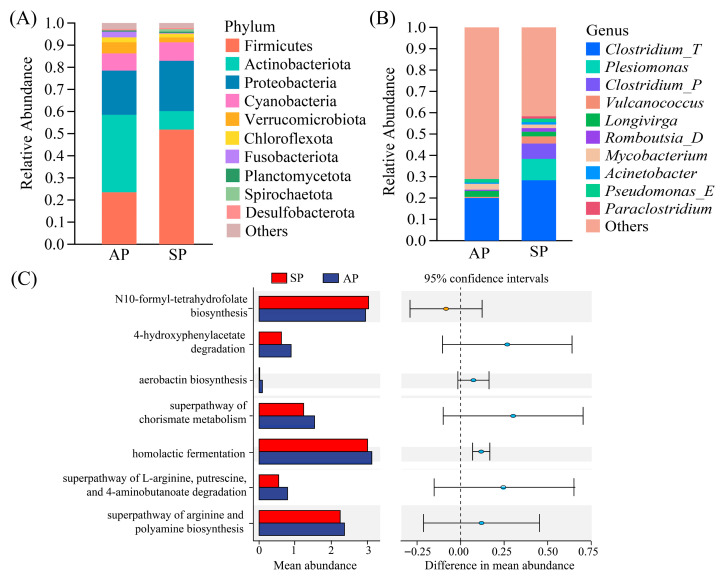 Figure 6
Figure 6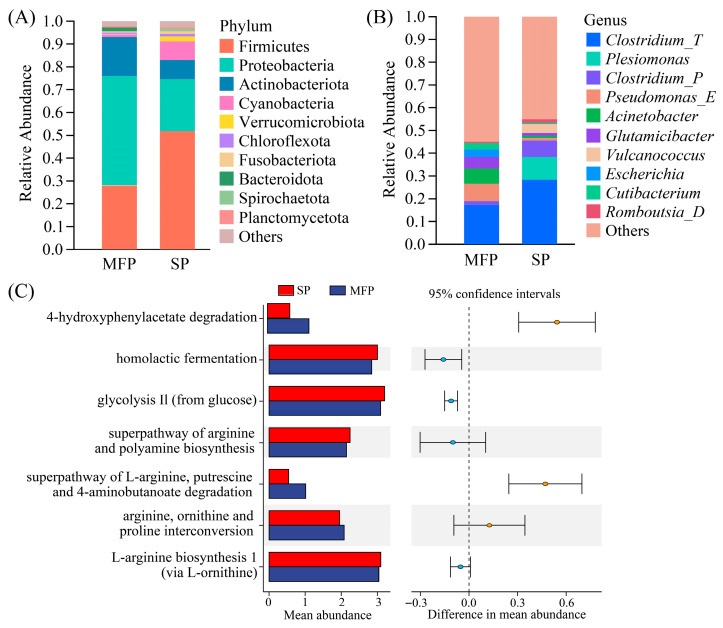 Figure 7
Figure 7- —National Key R&D Program of China
- —Monitoring of aquatic resources in key waters of Anhui province
- —Central Public-interest Scientific Institution Basal Research Fund, CAFS
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Community Ecology and Physiology · Gut microbiota and health · Aquaculture disease management and microbiota
1. Introduction
Coilia nasus (hereinafter referred to as C. nasus), belonging to the Engraulidae family within the Clupeiformes order, exhibits unique ecological adaptability that makes it an important model for studying fish resource conservation [1]. This species demonstrates two distinct ecological phenotypes: anadromous migration and freshwater residency. The former is extensively distributed across estuarine ecosystems along the northwest Pacific coast. During spring, mature individuals of this population exhibit a reproductive-driven bidirectional migration pattern between marine and freshwater environments, thereby developing a unique adaptive capability to salinity gradients [2]. Conversely, the latter primarily resides in lake ecosystems in the middle and lower reaches of the Yangtze River (e.g., Taihu Lake and Chaohu Lake), playing a crucial role in the regional biotic community by occupying a key ecological niche [3,4]. C. nasus has been classified as Endangered (EN) on the IUCN Red List of Threatened Species (www.iucnredlist.org, accessed on 25 February 2026), underscoring the critical and immediate need for conservation action.
In recent years, research into the composition, structure, and functional dynamics of host-intestinal microbiota interactions has advanced rapidly. The intestinal microbiome of fish serves as the “second genome” in host-environment interactions and plays a critical role in shaping the host’s nutritional metabolism and health homeostasis via pathways such as metabolic regulation and immune modulation [5,6,7,8]. Research demonstrates that the species-specific structure of the intestinal microbiota is collectively shaped by a variety of factors, including the host’s ecological lifestyle (migratory versus sedentary), geographical population distribution, developmental stages, and feeding strategies [9,10,11,12]. Compared with terrestrial vertebrates, the intestinal microbiota of fish exhibits pronounced dynamic variations and relatively lower biodiversity [13]. However, in widely distributed carnivorous species, discernible core microbiota are still present. These conserved microbial groups may harbor critical information regarding the coevolutionary processes between hosts and their associated microbiota [14,15,16,17].
Current research on C. nasus is predominantly concentrated in the Yangtze River estuary, its middle and lower reaches, and selected tributaries and connected lakes. Key research themes encompass population genetics [18]; age composition and somatic growth dynamics [19]; geographic population differentiation and classification [20,21]; stock assessment and demographic structure [22,23]; and integrative genomics—including functional annotation of differentially expressed genes and pathway enrichment analyses [24,25,26]. In contrast, investigations into the intestinal microbiota of C. nasus remain comparatively limited, with existing studies largely confined to intraspecific comparisons within a single ecotype (e.g., anadromous or freshwater-resident forms) [27,28]. Notably, a systematic, cross-ecotype comparative analysis of gut microbial community composition, diversity, and functional potential across distinct ecological groups of C. nasus is still lacking.
In recent years, driven by the rapid advancement of high-throughput sequencing technologies, 16S rRNA amplicon sequencing [29] has enabled high-resolution profiling of the intestinal microbiota. This study collected intestinal samples from three ecologically distinct populations of C. nasus: anadromous individuals (comprising both marine-phase and freshwater-phase migrants), sedentary individuals, and aquaculture-reared individuals. The primary objective was to establish a more comprehensive, resolved catalog of bacterial taxa inhabiting the gut microbiota of C. nasus. The study introduces three key conceptual and methodological advances: (1) the first systematic, multi-ecotype comparison of gut microbiota composition and predicted functional profiles across life history-divergent forms of C. nasus; (2) comparing the convergent and ecotype-specific patterns in gut microbiota structure across the anadromous, sedentary, and aquaculture-reared ecotypes of C. nasus; and (3) an integrative investigation linking inter-ecotype differences in microbiome structure and function to host-associated environment and dietary regimes, thereby advancing mechanistic understanding beyond mere correlation. These findings provide a novel theoretical framework for understanding host–microbe symbiosis in fish, while also offering a microbial ecological basis for formulating conservation and aquaculture strategies for C. nasus populations.
2. Materials and Methods
2.1. Sampling of C. nasus and Their Intestinal Tissues
To ensure comprehensive coverage of C. nasus sample types, our experimental group collected migratory marine population (MMP) of C. nasus from the East China Sea, migratory freshwater population (MFP) of C. nasus from the main stream of the Yangtze River and Poyang Lake, aquaculture-reared population (AP) of C. nasus from Yangzhong aquaculture bases and sedentary population (SP) of C. nasus from landlocked lakes. Figure 1 presents morphological illustrations of different ecological populations of C. nasus and Figure 2 illustrates sampling locations of different ecological populations of C. nasus. All individuals were confirmed as migratory C. nasus based on the Sr/Ca ratio analysis of otoliths (Figure S1). At each sampling site, 20 individuals of C. nasus were collected; morphometric and biological data were recorded for each individual. A total of 280 individuals were sampled across all ecological groups. Group-level statistics, including mean ± SD of total length and body weight, were then calculated separately for the anadromous, aquaculture-reared, and sedentary ecological populations (Table 1).
After measuring the biological indicators, C. nasus were immediately dissected on site. The external surface of each was sterilized with an ethanol-soaked cotton ball. Subsequently, the abdomen was carefully incised from the anus upward along the ventral ridge using scissors. The entire intestine was then isolated and transferred to a 2 mL cryotube. The samples were rapidly frozen in liquid nitrogen and subsequently stored at −80 °C in an ultra-low temperature freezer.
2.2. Extraction and Sequencing of Total Bacterial Genomic DNA
Total DNA from the intestinal microbiota was extracted using the E.Z.N.A.^®^ Soil DNA Kit (Omega Bio-Tek, Norcross, GA, USA, Cat. No. D5624-02). The quality of DNA was assessed by 1% agarose gel electrophoresis. The concentration and purity of DNA were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The V3–V4 variable region was amplified using the upstream primer (5′-CCTAYGGGRBGCASCAG-3′) and the downstream primer (5′-GGACTACNNGGGTATCTAAT-3′) [30]. Following PCR amplification, sequencing was conducted on the Illumina MiSeq platform (Illumina, San Diego, CA, USA), with three technical replicates established for consistency.
The PCR components involved Q5 reaction buffer (5×, 5 μL), Q5 High-Fidelity GC buffer (5×, 5 μL), Q5 High-Fidelity DNA Polymerase (5U/μL, 0.25 μL), each Forward and Reverse primer (10 μM, 1 μL), dNTPs (10 mM, 2 μL), DNA Template (2 μL), and ddH_2_O (8.75 μL). Thermal cycling consisted of initial denaturation at 98 °C for 5 min, followed by 25 cycles (denaturation at 98 °C for 30 s, annealing at 52 °C for 30 s, and extension at 72 °C for 45 s) with a final extension of 5 min at 72 °C. A total of PCR amplicons were purified with Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN, USA) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). After the individual quantification step, amplicons were pooled in equal amounts, and Single Molecule Real Time (SMRT) sequencing technology was performed using the PacBio Sequel platform at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China).
2.3. Bioinformatics Analysis of High-Throughput Sequencing Data
The primer fragments were removed from the sequences using Qiime’s cutadapt trim-paired, and sequences that did not match the primers were discarded. Subsequently, DADA2 was employed for sequence quality control, denoising, merging, and chimera removal. DADA2-based quality filtering was performed with the following parameters: truncLen = c(240, 200), maxN = 0, maxEE = c(2, 2), and truncQ = 2. Putative chimeric sequences were removed using the removeBimeraDenovo function (default settings). ASVs were taxonomically assigned to the species level using the greengenes2 reference database, which integrates phylogenetic frameworks from the Genome Taxonomy Database (GTDB) and refines broad bacterial taxonomic groups—such as the deeply branching lineages—into more granular, phylogenetically informed categories. After denoising, the feature sequences and ASV tables of ASVs (Amplicon Sequence Variants) were merged, and Singleton ASVs (ASVs with a total sequence count of only 1 across all samples) were removed. In the raw data obtained from high-throughput sequencing, an R script was used to analyze the length distribution of high-quality sequences in all samples, followed by ASV analysis on the final sequences.
Further analysis of species composition among samples was performed based on bacterial ASVs identified in the intestines. In this study, the relative abundance of each bacterium at a specific taxonomic level was visualized using bar charts, with particular attention given to the core microbiota of C. nasus. Astudillo-Garcia et al. proposed that defining the common core bacterial group (i.e., the bacterial community within the microbiome) requires setting both a detection threshold (relative abundance) and an existence rate percentage for bacterial classifications [31]. The range of common core annotations spans from as low as 30% [32] to 100% occurrence rates [33], while the detection threshold varies between 0.001% and 0.1%. In our study, bacterial ASVs present in over 50% of the same sample group were designated as part of the core microbiota (the core microbiota was defined as bacterial ASVs detected in at least 10 of the 20 individuals sampled from the same location).
A Venn diagram was employed to illustrate shared and unique bacterial species and their respective counts in the intestines of different C. nasus groups, and the contribution of each group’s intestinal microbiota to the total bacterial community was calculated (the contribution rate was calculated as the number of intestinal bacteria in each group of C. nasus divided by the total number of bacteria). The LEfSe method was utilized to identify robust differences in species between groups. The functional potential of the microbial communities was predicted using PICRUSt2 (v2.5.2) with the MetaCyc pathway database (v12.5) as the reference annotation resource.
3. Results and Analysis
3.1. Construction of the Background Database for Intestinal Microbiota in C. nasus
A total of 23,764,710 high-quality 16S rRNA gene sequences were obtained from intestinal samples of 280 individuals of C. nasus, with read lengths ranging from 235 to 446 base pairs (bp). These sequences were clustered into 33,371 prokaryotic amplicon sequence variants (ASVs). The dilution curves demonstrated that each curve became increasingly flat as the number of sequences increased, suggesting that the sequencing depth was adequate to capture the information of all microorganisms present in the samples (Figure S2). A total of 37 phyla, 106 classes, 280 orders, 525 families, 1144 genera, and 1707 species were identified in the intestinal samples of C. nasus. After averaging across the samples, the dominant bacterial taxa were statistically analyzed based on their relative abundances (Table S1). Among these, the top four dominant phyla collectively accounted for 92.20% of the total abundance, specifically Firmicutes, Proteobacteria, Actinobacteriota, and Cyanobacteria. In contrast, the top five dominant genera accounted for only 39.90% of the total abundance, namely Clostridium_T, Plesiomonas, Unclassified_f_Peptostreptococcaceae, Clostridium_P, and Pseudomonas_E.
3.2. Alpha and Beta Diversity Analysis of the Intestinal Microbiota of C. nasus
Alpha diversity indices were compared across the four ecological populations of C. nasus (MMP, MFP, AP, and SP) using the Kruskal–Wallis test, followed by Dunn’s post hoc test with Benjamini–Hochberg false discovery rate (FDR) correction for pairwise comparisons. Specifically, the Chao1 index was used to estimate species richness, whereas the Shannon and Simpson indices were applied to evaluate community diversity (Figure 3A). Significant differences in both microbial richness and alpha diversity were observed across the four ecological populations of C. nasus, with the marine-migratory (MMP) and freshwater-migratory (MFP) populations exhibiting the greatest divergence in intestinal microbiota composition (p < 0.05). Notably, the aquaculture-reared population (AP) displayed significantly higher microbial diversity and richness compared to the other groups (Table S2).
Inter-group beta diversity difference analysis among the four ecological populations of C. nasus revealed significant pairwise differences among all group comparisons (p < 0.01), as indicated by asterisks (*) (Figure 3B). Beta diversity analysis was conducted to examine the structural composition of intestinal microbial communities across ecological types (Figure 3C). Non-metric multidimensional scaling (NMDS) was utilized to visualize community relationships, where shorter intergroup distances indicate greater similarity in microbial assemblages. The NMDS plot revealed clear separation among the four ecological groups, with only minor overlap, indicating distinct microbial community structures across groups. Among these, the migratory freshwater population (MFP) exhibited the highest degree of dispersion, followed by the sedentary population (SP), whereas the aquaculture-reared population (AP) showed the least variation, suggesting greater homogeneity in its intestinal microbiota.
3.3. The Structure of the Intestinal Microbiota in Different Ecological Populations of C. nasus
A petal diagram was employed to visualize community composition and identify group-specific species across the four ecological populations of C. nasus. At the phylum level (Figure 4A), the four ecological groups of C. nasus shared 20 bacterial phyla. The migratory marine population (MMP) was identified with 21 bacterial phyla, contributing 56.76% to the total bacterial phyla of C. nasus. The migratory freshwater population (MFP) was identified with 30 bacterial phyla, contributing 81.08%. Similarly, the aquaculture-reared population (AP) contained 30 bacterial phyla, with Elusimicrobiota being its unique phylum. The sedentary population (SP) was identified with 35 bacterial phyla, contributing as high as 94.59%, and Campylobacterota, WOR-3, and Desulfobacterota_E were exclusively present in this group. Similarly, at the genus level (Figure 4B), the four ecological groups of C. nasus shared 127 bacterial genera. Specifically, the MMP harbored 259 bacterial genera, contributing only 22.64% to the total bacterial genera, with 31 unique genera. The MFP contained 592 bacterial genera, contributing 51.75%. The AP had 533 bacterial genera, contributing 46.59%. Lastly, the SP harbored 844 bacterial genera, contributing 73.78% (Table S4).
To compare the differences in the relative abundance of intestinal microbiota among different ecological groups of C. nasus, this study tallied the top 10 species compositions at the phylum and genus levels, respectively. At the phylum level (Figure 4C), Proteobacteria, Firmicutes, and Actinobacteriota were the dominant bacterial phyla in C. nasus, accounting for 78.81%~94.99% of the total abundance. Notably, the relative abundance of Firmicutes in the intestinal microbiota of the SP was higher than that in other ecological types of C. nasus. At the genus level (Figure 4D), bacterial groups with lower abundance rankings accounted for a significantly larger proportion, contrasting with observations at the phylum level. Notably, Photobacterium exhibited much higher relative abundance in the intestinal microbiota of the MMP C. nasus compared to other ecological types. Clostridium_T showed relatively high abundance in the MFP, AP, and SP of C. nasus. Additionally, Plesiomonas was markedly more abundant in the sedentary group than in other ecological types of C. nasus.
3.4. The Intestinal Microbiota of Migratory C. nasus Varies Under Different Living Environments
The intestinal microbiota of anadromous C. nasus, which migrate from the ocean to the Yangtze River (freshwater environment) for reproductive migration, exhibit significant differences due to variations in external environmental factors, including salinity and chemical composition. From the perspective of species composition, at the phylum level (Figure 5A), the dominant bacterial phyla in both migratory marine (MMP) and freshwater populations (MFP) of C. nasus were Proteobacteria, Actinobacteriota and Firmicutes, collectively accounting for 93.46% to 94.99%. At the genus level (Figure 5B), the dominant bacterial genera in marine-migratory C. nasus were Photobacterium, Psychrobacter, and Aliivibrio, whereas those in the MFP C. nasus were Clostridium_T, Pseudomonas_E, and Acinetobacter.
Further analysis of the metabolic pathways exhibiting significant differences between the MMP and the MFP of C. nasus revealed that the MMP was enriched in aerobactin biosynthesis and arginine, ornithine, and proline interconversion pathways. While the MFP of C. nasus was enriched in the pathways of 4-hydroxyphenylacetate degradation, superpathway of chorismate metabolism, and superpathway of L-arginine, putrescine, and 4-aminobutanoate degradation (Figure 5C).
3.5. The Intestinal Microbiota Composition Differences Between Aquaculture-Reared and Sedentary Population C. nasus
Both aquaculture-reared population (AP) and sedentary population (SP) C. nasus do not migrate; however, their food sources differ markedly, which influences the composition of their intestinal microflora. At the phylum level (Figure 6A), the dominant bacterial phyla in both groups are Firmicutes, Actinobacteriota, Proteobacteria, and Cyanobacteria, accounting for 86.16% to 90.84% of the total abundance. Notably, the relative abundance of Actinobacteriota is significantly higher in the AP C. nasus than in the SP C. nasus (35.05% vs. 8.37%). While at the genus level (Figure 6B), Clostridium_T was the dominant genus in both the AP and the SP C. nasus, accounting for 19.99% to 28.33% of the total abundance. Notably, the relative abundance of Plesiomonas was markedly higher in the SP C. nasus than in the AP C. nasus (10.28% vs. 0.02%).
Similarly, we performed a statistical analysis of the differential metabolic pathways between the AP and the SP of C. nasus (Figure 6C). The results showed that in the AP C. nasus, the enriched pathways included 4-hydroxyphenylacetate degradation and the superpathway of chorismate metabolism, whereas in the SP C. nasus, the enriched pathway was N10-formyl-tetrahydrofolate biosynthesis.
3.6. The Intestinal Microbiota of Migratory Freshwater and Sedentary Population C. nasus Differ Significantly
The migratory freshwater population (MFP) and the sedentary population (SP) C. nasus exhibit marked differences in life habits, reproductive behaviors, and physiological adaptations. Based on these distinctions, we aimed to characterize their intestinal microbiota. At the phylum level (Figure 7A), Proteobacteria, Firmicutes, and Actinobacteriota were the dominant phyla in both groups, accounting for 80.04% to 89.32% of the total abundance. Notably, the relative abundance of Cyanobacteria was substantially higher in the SP C. nasus than in the MFP (8.22% vs. 1.07%). At the genus level (Figure 7B), Clostridium_T was the dominant genus in both the MFP and the SP C. nasus, accounting for 17.13% to 28.33% of the total abundance. The relative abundances of Pseudomonas_E (7.83% vs. 1.29%), Acinetobacter (6.62% vs. 1.44%), and Glutamicibacter (4.95% vs. 0.68%) were significantly higher in the MFP C. nasus than in the SP ones. In contrast, the relative abundances of Plesiomonas (10.28% vs. 0.17%) and Clostridium_P (6.72% vs. 1.73%) were markedly higher in the SP C. nasus than in the MFP ones.
The metabolic pathways of the intestinal microbiota in the MFP and the SP C. nasus differed significantly (Figure 7C). In the MFP, the enriched pathways included 4-hydroxyphenylacetate degradation, the superpathway of L-arginine, putrescine, and 4-aminobutanoate degradation, as well as arginine, ornithine, and proline interconversion. By contrast, in the SP C. nasus, the enriched pathways were homolactic fermentation, glycolysis III (from glucose), and the superpathway of arginine and polyamine biosynthesis.
4. Discussion
4.1. Overview of the Intestinal Microbiota in C. nasus
The development of high-throughput sequencing technology has enabled comprehensive and efficient collection and analysis of intestinal microbiota data, thereby enhancing our understanding of the structure and function of the intestinal flora [34]. The primary objective of this research was to establish a more comprehensive, resolved catalog of bacterial taxa inhabiting the gut microbiota of C. nasus, and to characterize microbiome features linked to the integrated “life history–habitat–diet” model across its major ecotypes. A stratified sampling strategy was implemented to collect 280 individuals of C. nasus from 14 geographically distinct sampling sites, representing three ecotypes: migratory (comprising marine and freshwater subpopulations), sedentary, and aquaculture-reared. Intestinal luminal contents were then subjected to high-throughput 16S rRNA gene amplicon sequencing. A total of 37 phyla, 1144 genera, and 1707 species were identified in the sequencing results, demonstrating that the intestinal microbiota of C. nasus exhibits high diversity and occupies a relatively complex microbial ecological niche.
Species annotation revealed that Firmicutes, Proteobacteria, Actinobacteriota, and Cyanobacteria are the predominant bacterial phyla in the gut of C. nasus. Nie et al. employed PCR-DGGE fingerprinting technology to analyze the bacterial community structures in wild Yangtze River C. nasus and aquaculture-reared C. nasus [35,36]. Their results revealed that the dominant bacterial phyla in pre-migratory juveniles and post-migratory adults of wild C. nasus were Proteobacteria, Actinobacteria, and Firmicutes, whereas Proteobacteria and Actinobacteria predominated in aquaculture-reared C. nasus. This study significantly improved species resolution by leveraging a larger sample size and second-generation sequencing technology. It not only validated previous findings but also identified novel dominant phyla. Specifically, Firmicutes are involved in carbohydrate fermentation and produce short-chain fatty acids such as butyrate, which helps maintain intestinal barrier function and plays an essential role in fish health [37]. Proteobacteria contribute to maintaining the homeostasis of the intestinal anaerobic environment [38] and provide absorbable monosaccharide substrates for fish through the degradation of polysaccharides (e.g., cellulose and galactose) [39]. Actinobacteriota play a critical role in defending against pathogen invasion in C. nasus by producing antibiotics and secondary metabolites [40].
Similarly, the research results indicate that the dominant bacterial genera in C. nasus include Clostridium_T, Plesiomonas, Clostridium_P, and Pseudomonas_E. A comparison with previous studies reveals significant variations in the dominant bacterial genera among C. nasus of different ecological types. For example, Yang et al. identified Rhodococcus and Photobacterium as the predominant genera in the intestinal microbiota of C. nasus from the Yangtze River Estuary [28]. Duan et al. reported that the gut microbiota of C. nasus from Taihu Lake is primarily composed of Bacteroides, Faecalibacterium, Halomonas, and Mycobacterium [41]. Li et al. demonstrated that the gut microbiota of Coilia nasus from four water bodies—the mainstream of the Yangtze River (Pengze section), Poyang Lake, Qingcaosha Reservoir, and Shengsi Sea—is predominantly characterized by Clostridium, Phocaeicola, Psychrobacter, Ralstonia, Acinetobacter, and Bacteroides [42].
The composition of dominant bacterial genera exhibits significant variation across different ecological types of C. nasus. Mandal et al. demonstrated that a substantial proportion of microorganisms in fish are acquired from the water, food, and sediment in their growth environment [43]. Consequently, even within the same ecological type of C. nasus, intestinal microbiota can exhibit significant variation due to differences in developmental stage, diet, and collection basin. Specifically, members of Clostridium are frequently involved in polysaccharide degradation and short-chain fatty acid (SCFA) synthesis [44], whereas Plesiomonas can influence intestinal health by modulating the host immune response [45]. These findings suggest that the aforementioned bacterial genera may play a central role in the host’s nutritional metabolism.
Fish of different ecological types, having undergone long-term adaptation to distinct environmental conditions—such as salinity, temperature, diet, and migratory stress—exhibit significant differentiation in the diversity and structure of their intestinal microbiota. This divergence reflects the combined effects of host–microbe co-adaptation and environmental filtering [46,47,48]. The alpha diversity of the intestinal microbiota in the aquaculture-reared population (AP) and sedentary population (SP) of C. nasus is significantly higher than that in the anadromous (river–sea migratory) group. Furthermore, microbial variation among AP is the smallest, likely due to the relatively stable aquaculture environment and freshwater habitats (e.g., lakes), where food resources are more consistent and diverse. Such conditions provide a broader range of nutritional substrates and a stable niche for microbial colonization, thereby promoting the establishment and persistence of a more diverse gut microbiota. This observation is consistent with findings from Deng’s study on wild and farmed Coreius guichenoti, which similarly demonstrated higher microbial diversity in captive populations [49]. The Simpson diversity index shows minimal fluctuation, suggesting high community evenness and relative stability of dominant amplicon sequence variants (ASVs). Nevertheless, alpha diversity is generally dynamic across both short- and long-term temporal scales [50].
4.2. Salinity Significantly Influences the Composition of Intestinal Microbiota in Migratory Marine and Freshwater Populations of C. nasus
The salinity difference between seawater and freshwater is highly pronounced. The intestinal microbiota diversity in the migratory marine population (MMP) of C. nasus is lower compared to that in the freshwater population, potentially due to the inhibitory effects of high salinity on certain bacterial groups [27]. Elevated salinity imposes osmotic stress that inhibits the growth of freshwater and euryhaline microorganisms, thereby driving compositional shifts in microbial community structure [51]. Cui et al. discovered that the high salinity of seawater may suppress the growth of some obligate anaerobic bacteria (e.g., Bacteroides) [52], leading to slightly reduced bacterial diversity compared to freshwater fish.
In terms of species composition, Photobacterium and Psychrobacter were significantly enriched in the intestines of the MMP C. nasus. Notably, Photobacterium is a common luminescent bacterial genus found in marine fish [53]. Silva et al. previously identified Photobacterium spp. in the stomach and intestinal samples of Lateolabrax japonicas [54]. Its metabolic functions include salt ion regulation and lipid decomposition [55,56]. Fan et al. demonstrated that Photobacterium participates in the metabolism of carbohydrates and amino acids in the gut microbiota of Hexagrammos otakii [57]. The majority of Psychrobacter species have been isolated from various cold marine environments [58]. Studies indicate that Psychrobacter is a symbiotic member of the gastrointestinal microbiota in marine fish such as Paralichthys adspersus, Oreochromis mossambicus, and Gadus morhua [59,60,61]. Similarly, the relative abundance of Clostridium_T in the migratory freshwater population (MFP) of C. nasus was significantly higher than that in the MMP. Notably, common strains such as Clostridium_sensu_stricto_1 dominate the intestines of juvenile Acipenser dabryanus, comprising over 70% of the anaerobic bacteria in their intestines [62]. This strain plays a critical role in promoting the production of short-chain fatty acids (SCFAs) and enhancing the structure of intestinal microbiota [63].
Moreover, the MMP of C. nasus exhibits enrichment of genes associated with aerobactin biosynthesis. Upon migrating into the sea, this population enters the fattening stage, during which feeding intensity markedly increases [64]. Consequently, the enrichment of this pathway may be associated with the elevated energy demands required for nearshore development in the migratory marine population of C. nasus. In contrast, pathways such as L-arginine degradation and 4-hydroxyphenylacetic acid degradation are enriched in the MFP of C. nasus, potentially reflecting its adaptation to fluctuating energy demands. During freshwater migration, C. nasus exhibits minimal feeding activity and relies on the conversion of non-sugar substances to sustain basic physiological functions [65].
4.3. The Dietary Composition Significantly Influences the Intestinal Microbiota Structure of Aquaculture-Reared and Sedentary Population C. nasus
Both aquaculture-reared population (AP) and sedentary population (SP) C. nasus populations are non-migratory and maintain stable food sources. However, a significant difference was observed in the dietary composition between the two groups. To ensure accelerated growth, the AP C. nasus are fed a combination of live prey—including cladocerans, copepods, and shrimp post-larvae—supplemented with nutritionally balanced artificial compound feed [66]. In contrast, the SP C. nasus primarily consume planktonic organisms like copepods and cladocerans, along with small aquatic animals [67]. The unique phylum Elusimicrobiota, detected in the SP C. nasus, is frequently observed in the intestines of ruminants [68]. Zhao et al. identified in their study on the intestinal microbiome of Sus scrofa domesticus that Elusimicrobiota plays a role in the synthesis of essential amino acids and vitamins [69]. Under aquaculture conditions, where diets are often monotonous, hosts may depend on microbial-assisted metabolism for nutrient acquisition [70]. The inclusion of plant fibers in artificial feed could potentially promote the colonization of this bacterial group. The abundance of Plesiomonas in the SP C. nasus is significantly elevated relative to other ecological communities. Prior evidence indicates that Plesiomonas (e.g., P. shigelloides) is detected in freshwater fish and may propagate indirectly through aquatic food webs [71]. Therefore, the elevated abundance of Plesiomonas in the intestines of sedentary C. nasus is tentatively associated with their zooplankton-dominated diet, while explicitly acknowledging that this inference lacks empirical support from direct dietary data.
The comparison of metabolic pathways reveals that the AP C. nasus exhibit enrichment in the 4-hydroxyphenylacetic acid degradation pathway, potentially linked to phenolic compounds present in artificial feed [72,73]. Phenolic substances promote digestive health and nutrient utilization by modulating the intestinal microbiota composition—specifically enriching beneficial bacterial populations—which enhances nutrient absorption and suppresses intestinal pathogens [74,75]. In contrast, the folate biosynthesis pathway enriched in the SP C. nasus may support their reproductive requirements under lower-nutrient conditions [76]. The pronounced differences in intestinal microbiota between the AP and the SP C. nasus underscore the significant impact of dietary sources.
4.4. Life History Strategies Shape the Intestinal Microbiota Communities of Migratory Freshwater and Sedentary Population C. nasus
Migratory C. nasus experience prolonged energy expenditure during migration and feed minimally during this period, whereas sedentary C. nasus exhibit lower movement intensity and have a more stable food supply. These differing life history strategies are the primary drivers of the distinct intestinal microbiota compositions observed in these two populations [77]. The number of bacterial phyla and genera in sedentary population (SP) C. nasus is significantly higher than that in migratory freshwater population (MFP) C. nasus. The unique phylum Campylobacterota found in the SP C. nasus correlates with specific organic matter conditions in the environment [78]. The enrichment of Pseudomonas_E in the MFP of C. nasus may enhance the host’s metabolic flexibility in variable environments. For instance, Ramasam et al. reported that different strains of Pseudomonas exhibit varying metabolic capabilities across diverse carbon sources [79]. The SP C. nasus inhabit lake ecosystems, where the high abundance of Cyanobacteria may be linked to the frequent occurrence of cyanobacterial blooms in their habitats (e.g., Taihu Lake and Chaohu Lake) [80]. This bacterial community can provide additional nitrogen sources for the host via nitrogen fixation [81].
The differences in metabolic pathways further substantiate the aforementioned hypothesis. The amino acid degradation pathway is potentially linked to energy metabolism in the MFP of C. nasus [65]. In contrast, glycolysis may serve as a key metabolic hub supporting the biosynthesis of secondary metabolites in the SP C. nasus, thereby augmenting its competitive fitness under stable environmental conditions [82]. The microbial distinctions between the anadromous freshwater population and the SP C. nasus underscore the influence of the host’s life history strategy on microbiota composition.
5. Conclusions
This study utilized high-throughput sequencing technology to analyze the compositional characteristics and metabolic functional differences in intestinal microbiota in four ecological groups of C. nasus (migratory marine, migratory freshwater, sedentary and aquaculture-reared populations), further elucidating the ecological adaptation mechanisms of host–microbiota interactions under varying life history strategies. A relatively comprehensive background database of C. nasus intestinal microbiota was established. Results demonstrated that the composition and function of C. nasus intestinal microbiota were significantly influenced by living environments and dietary sources, while migratory behavior also played a role in shaping distinct microbiota structures and metabolic features. The succession of metabolic functions in intestinal microbiota corroborated these findings.
These insights provide critical evidence for understanding the symbiotic mechanisms of fish–host–microbiota interactions. The research outcomes hold substantial significance for enhancing the background database of C. nasus intestinal microbiota and offer important implications for distinguishing C. nasus origins via intestinal microbiota analysis. Additionally, this study lays a foundation for future investigations into C. nasus microbiota ecology. However, PICRUSt2-based functional inference is inherently constrained by the phylogenetic breadth and functional completeness of its underlying reference genome database, introducing potential biases in predicted metabolic profiles. To address these limitations, predicted metabolic functions will be experimentally validated using shotgun metagenomic sequencing and untargeted metabolomics. Moreover, the precise mechanistic links between diet and the intestinal microbiota of C. nasus remain incompletely understood. Future studies should prioritize controlled comparative analyses of the gut microbiota in wild and farmed populations, integrating targeted dietary interventions to rigorously assess how specific nutritional components shape microbial community assembly, functional potential, and host-associated phenotypes. Such efforts hold translational promise for steering the microbiota of farmed C. nasus toward configurations characteristic of wild conspecifics, thereby supporting improved physiological homeostasis and environmental resilience.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hong Z.Z. Mei X.L. Wang M.M. Zhang Z.M. Wang W.W. A review on the biological characteristics and resource protection status of Coilia nasus in the Yangtze River basin Aquaculture 202344364110.3969/j.issn.1004-2091.2023.07.007 · doi ↗
- 2Zhu C. Yang P. Migration biology and stress response of Coilia nasus: A review Chin. Agric. Sci. Bull.20233913013410.11924/j.issn.1000-6850.casb 2021-1070 · doi ↗
- 3Cheng W.X. Tang W.Q. Some phenotypic varieties between different ecotypes of Coilia nasus in Yangtze River Chin. J. Zool.201146334010.1111/j.1749-4877.2010.00232.x · doi ↗
- 4Xu G.C. Gu R.B. Zhang C.X. Zheng J.L. Wen H.B. Xu P. Comparison and quality evaluation of nutritional composition in two ecological groups of Coilia nasus: “Jiangdao” (river ecotype) and “Haidao” (marine ecotype)Mar. Fish.20093138739510.3969/j.issn.1004-2490.2009.04.010 · doi ↗
- 5Clements K.D. Angert E.R. Montgomery W.L. Choat J.H. Intestinal microbiota in fishes: What’s known and what’s not Mol. Ecol.2014231891189810.1111/mec.1269924612310 · doi ↗ · pubmed ↗
- 6Lee W.J. Hase K.J. Gut microbiota-generated metabolites in animal health and disease Nat. Chem. Biol.20141041642410.1038/nchembio.153524838170 · doi ↗ · pubmed ↗
- 7Ley R.E. Hamady M. Lozupone C. Turnbaugh P.J. Ramey R.R. Bircher J.S. Lozupone C. Schlegel M.L. Tucker T.A. Schrenzel M.D. Evolution of mammals and their gut microbes Science 20083201647165110.1126/science.115572518497261 PMC 2649005 · doi ↗ · pubmed ↗
- 8Mckenney E.A. O’Connell T.M. Rodrigo A. Yoder A.D. Feeding strategy shapes gut metagenomic enrichment and functional specialization in captive lemurs Gut Microbes 2018920221710.1080/19490976.2017.140876229182421 PMC 6219591 · doi ↗ · pubmed ↗