Antipsychotic Medications in Parkinson’s Disease Psychosis; A Systematic Review of Double-Blind, Randomised, Placebo-Controlled Trials
Christopher John McKeown, Alberto Salmoiraghi

TL;DR
This review finds that clozapine is effective for Parkinson’s disease psychosis, while quetiapine lacks strong evidence for its use.
Contribution
The study systematically evaluates antipsychotic medications for Parkinson’s disease psychosis using randomized, placebo-controlled trials.
Findings
Clozapine improves Parkinson’s disease psychosis with large effect sizes at low doses and does not worsen motor function.
Quetiapine has limited evidence for treating Parkinson’s disease psychosis despite frequent clinical use.
Pimavanserin also shows significant improvement in psychosis symptoms without worsening motor scores.
Abstract
Main findings Clozapine shown to improve Parkinson’s disease psychosis (PDP) with a large effect size despite low doses and does not appear to worsen motor function.Quetiapine has a poor evidence base for treatment of PDP, despite widespread use. Clozapine shown to improve Parkinson’s disease psychosis (PDP) with a large effect size despite low doses and does not appear to worsen motor function. Quetiapine has a poor evidence base for treatment of PDP, despite widespread use. Implications Clozapine could be considered as a first line antipsychotic (AP) for the management of PDP if standard reductions in dopaminergic medications fail to achieve resolution of symptoms.APs with low Dopamine receptor 2 (D2) affinity may not appreciably worsen motor symptoms of Parkinson’s Disease. Clozapine could be considered as a first line antipsychotic (AP) for the management of PDP if standard…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
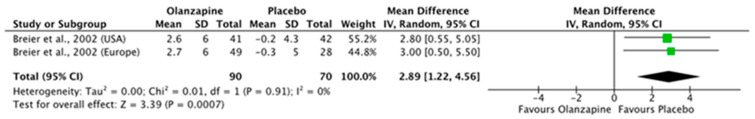 Figure 1
Figure 1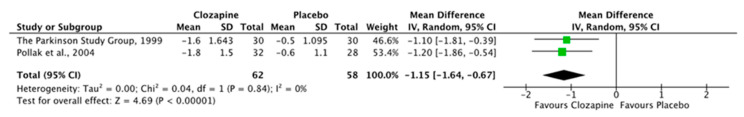 Figure 2
Figure 2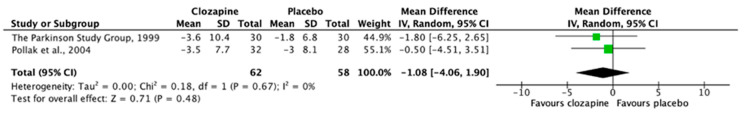 Figure 3
Figure 3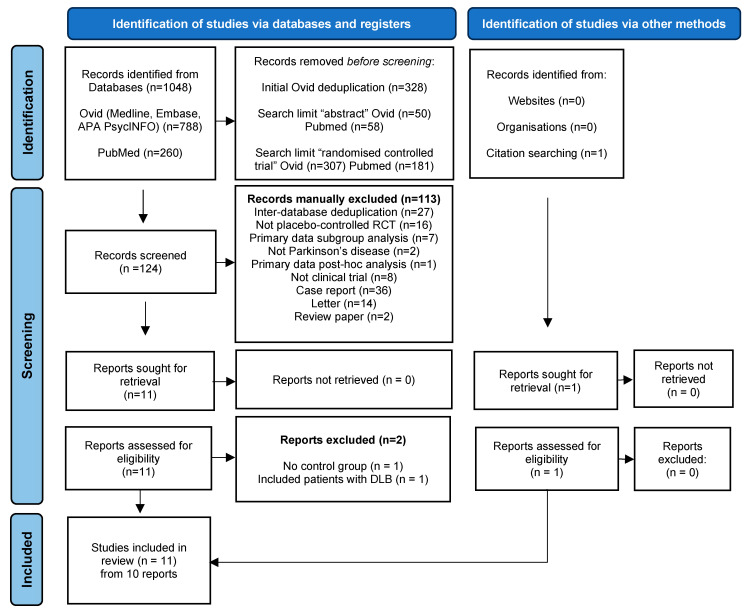 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6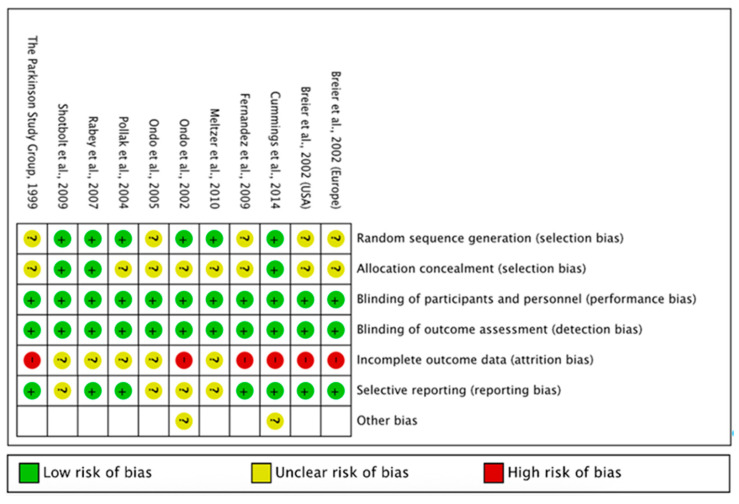 Figure 7
Figure 7| Author | Psychosis Outcomes | Motor Outcomes | Dropouts | Adverse Events | ||
|---|---|---|---|---|---|---|
| Active | Control | Active | Control | |||
| Shotbolt et al., 2009 | No statistically significant changes in psychosis between groups as measured by NPI, Baylor scale and BPRS. | UPDRS III (motor) scores not significantly different between groups | Participants N = 11 | Participants N = 13 | 3 drowsiness | 2 drowsiness |
| Rabey et al., 2007 | No significant changes in psychosis from baseline and between both groups, as measured with BPRS and CGI-S. | UPDRS III (motor) scores not significantly different between groups | Participants N = 30 | Participants N = 28 | 7—Somnolence | 2—Somnolence |
| Ondo et al., 2005 | No significant changes in psychosis between groups as measured by BPRS and Baylor scale. | UPDRS II (Activities of daily living) + III (motor) scores + GDRS not significantly different between groups | Participants N = 21 | Participants N = 10 | 9—Sedation | 4—Sedation |
| Fernandez et al., 2009 | Statistically significant improvement in psychosis in Quetiapine group vs. placebo as measured by BPRS item 12 (Hallucinations) and CGI-S. | UPDRS III (motor) scores not significantly different between groups | Participants N = 8 | Participants N = 8 | 1—Confusion | 1—Bronchitis |
| Ondo et al., 2002 | No significant changes in psychosis between groups as measured by structured interview for hallucinations. | UPDRS III (motor) scores significantly worsened in active group compared with placebo. | Participants N = 18 | Participants N = 12 | 6—Worsening movement | 1—Insomnia |
| Breier et al., 2002 | No significant changes in psychosis between groups as measured by positive symptoms cluster sub-score of BPRS. | UPDRS total, II (ADL), III (motor) scores significantly worsened in Olanzapine group compared with placebo. | Participants N = 41 | Participants N = 42 | 10—Extrapyramidal syndrome | 1—Extrapyramidal syndrome |
| Breier et al., 2002 | No significant changes in psychosis between groups as measured by positive symptoms cluster sub-score of BPRS | UPDRS total, II (ADL), III (motor) scores significantly worsened in active group compared with placebo. | Participants N = 49 | Participants N = 28 | Not detailed | Not detailed |
| The Parkinson Study Group, 1999 | Statistically significant improvement in psychosis in Clozapine group compared with placebo as measured by BPRS, BPRS-M, CGI-S, SAPS. | UPDRS total, III (motor) scores not significantly different between groups. | Participants N = 30 | Participants N = 30 | Not detailed | Not detailed |
| Pollak et al., 2004 | Statistically significant improvement in psychosis in Clozapine group compared with placebo as measured by CGI-S and positive PANSS | No statistical worsening of motor scores between groups | Participants N = 32 | Participants N = 28 | 7—Worsening of PD | 1—Worsening of PD |
| Cummings et al., 2014 | Statistically significant improvement in psychosis in Pimavanserin group compared with placebo as measured by SAPS-PD | UPDRS II, III + composite score all showed no statistically significant worsening of motor scores between groups. | Participants N = 105 | Participants N = 95 | 6—Nausea | 6—Nausea |
| Meltzer et al., 2010 | No significant changes in psychosis between groups as measured by SAPS (Primary outcome measure) | UPDRS II, III + composite score all showed no statistically significant worsening of motor scores between groups. | Participants N = 29 | Participants N = 31 | 3—Somnolence | 5—Hallucinations |
| Receptor | Clozapine 1 | Quetiapine 1 | Olanzapine 1 | Pimavanserin 2 |
|---|---|---|---|---|
| α1A | 1.62 | 22 | 112 | nr |
| α2A | 37 | 3630 | 314 | nr |
| α2C | 6 | 28.85 | 28.9 | nr |
| D2 | 157 | 379 | 34.23 | nr |
| D3 | 269.1 | 340 | 47 | nr |
| H1 | 1.13 | 6.9 | 2.19 | nr |
| M1 | 6.17 | 489 | 2.5 | nr |
| 5-HT1A | 123.7 | 394.2 | 2282 | nr |
| 5-HT2A | 5.35 | 912 | 3.73 | 0.4 |
| 5-HT2C | 9.44 | 1843 | 10.2 | 1.6 |
| 5-HT6 | 13.5 | 948.75 | 8.07 | nr |
| 5-HT7 | 17.95 | 307.6 | 105.2 | nr |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsParkinson's Disease Mechanisms and Treatments · Parkinson's Disease and Spinal Disorders · Neurological disorders and treatments
1. Introduction
Parkinson’s Disease (PD) is a progressive, neurodegenerative disorder characterised by bradykinesia, rigidity, resting tremor and postural instability, resulting from neuronal cell loss within the Substantia Nigra Pars Compacta and subsequent dopamine (DA) depletion [1]. It is the second most common neurodegenerative disorder globally, affecting over 6 million individuals [2], with the fastest growing prevalence; incident cases exceeding one million annually as of 2017 [3,4]. Although diagnostic criteria focus on motor dysfunction, PD also presents with other non-motor symptoms, which may occur up to 20 years prior to clinical motor manifestation [5]. This prodromal phase of PD can be characterised by autonomic disturbance, olfactory dysfunction, sleep disorders and psychiatric symptoms [6,7,8,9,10].
Psychotic symptoms may occur in up to 75% of PD patients during the course of the disease process, depending on psychosis definitions and rating scales [11,12,13,14], and are a significant burden of disease. Parkinson’s disease psychosis (PDP) is associated with increased morbidity, depression, poorer quality of life [15], caregiver burden [16], development and progression of dementia [17,18] and has been shown to be a strong predictive factor for long-term care placement [19]. Recent studies show those diagnosed with PDP at an increased risk of hospitalisation [20] and up to 71% increase in risk of mortality compared with PD patients free from psychotic symptoms [21].
Dopaminergic medications have long been suggested as the main causative factor leading to PDP [22,23], with all PD medication types associated with psychotic symptoms [24]. Changes in dopaminergic therapy have been observed to precipitate PDP, with improvements in psychosis seen following reduction or cessation [25], however psychotic symptoms have been reported in up to 42% of drug-naive PD patients [26,27], suggesting that their occurrence may be intrinsically linked with the disease process itself rather than solely attributable to dopaminergic therapy used to manage motor dysfunction.
When psychotic symptoms are distressing, intolerable or fail to improve with adjustment of dopaminergic therapy, AP treatment may be required [28]. The use of AP medications in patients with PD is widespread, with 12% of patients prescribed an AP within the first 12 months of PD diagnosis and 35% of patients within seven years [29,30], and has been associated with worsened motor symptoms of PD and increased mortality [31].
Currently, only clozapine has a UK licence for the treatment of PDP, however accounts for <2% of AP prescribing. Quetiapine accounts for 49–66% of AP prescriptions in PDP [29,32], despite no clear advantage over placebo in a previous meta-analysis of double-blind, randomised placebo-controlled trials [33].
Previous systematic reviews and meta-analyses investigating APs in PDP have been completed, with a number limited to atypical class [34,35,36,37,38,39] or single drug reviews [33,40]. A single meta-analysis [34] completed in 2015 includes wide search terms for APs, and specifies only double blind, randomised, placebo-controlled trials for inclusion, and further systematic reviews and network meta-analyses have been completed including any APs whilst also including comparator [37] and open label studies [36,39]. Details of relevant studies are summarised on Table 1.
This review aims to provide an up-to-date analysis of double-blind, randomised, placebo-controlled trials investigating the treatment of PDP with no restriction by AP class, searching all AP medications that are in use or have been used, worldwide. The most recent systematic review with these strict inclusion criteria was published in 2015 [34] and this review serves as an appropriate update to the current body of literature. Preliminary findings from this review were presented in abstract and poster form at the Royal College of Psychiatrists International Congress, 2024. This manuscript has been substantially expanded and updated with recent literature searches, detailed methodology including risk of bias assessment, reporting of dose titration schedules and how these may have impacted outcomes, with a focus on detailed narrative synthesis to help inform readers of the strengths and weaknesses of the included studies. A limited pooled meta-analysis was also performed to provide supporting quantitative evidence. This review aims to further inform clinical decision-making regarding AP choice, dosing and titration schedules in PDP, given ongoing uncertainty about the relative roles of commonly used agents in routine practice, where quetiapine often remains a first-line treatment.
2. Materials and Methods
2.1. Study Design
This systematic review was conducted with reference to the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) 2020 statement guidelines (for PRISMA flow diagram, see Appendix A, Figure A1) [41]. The review protocol was developed a priori, however was not registered.
2.2. Objectives
The aims of this review are to identify, appraise and synthesise evidence from published, peer-reviewed double-blind, randomised, placebo-controlled trials investigating the efficacy and tolerability of APs in the management of PDP, with particular focus on motor outcomes.
2.3. Search Strategy
Searches were restricted to fully published and peer-reviewed papers: Electronic database searches were conducted via Ovid, including MEDLINE (1946 to 7 November 2025), Embase (1974 to 2025 Week 45), APA PsycINFO (1806 to Nov 2025 Week 1). Additional searches were performed in PubMed and Google Scholar to identify further relevant references (Nov 2025). Clinical trial registries, unpublished and non-peer reviewed sources were not systematically searched or included. Peer-review was considered an essential criterion for study inclusion, to ensure methodological scrutiny. The list of APs was derived by searching available literature and the online resources [42,43,44]. To maximise yield of relevant papers, selected keywords were truncated to account for differences in spelling and pluralisation and to cover a broader range of relevant related terms. Search strategy for each database can be found in Appendix A, Figure A2a,b.
2.4. Eligibility
Inclusion Criteria:
- Double-blind, randomised, placebo-controlled trials.
- Treatment arm using recognised AP medication.
- Participants with a clinical diagnosis of idiopathic PD and psychotic symptoms
- Studies reporting quantified measures of psychosis and motor outcomes using validated rating scales as either primary or secondary outcome measure.
- English-language, peer reviewed publications.
Exclusion Criteria:
- Comparator or open-label studies.
- Reviews, case reports, conference abstracts without full data or unpublished data.
- Studies including patients with diagnosed dementias such as dementia with Lewy bodies, unless data could be separated.
2.5. Outcomes of Interest
- Primary Outcome: Change in psychotic symptoms in PD, measured by validated instruments.
- Secondary outcome: Change in Motor symptoms, measured using validated instruments
- Adverse events and discontinuations reported.
2.6. Study Selection and Data Extraction
Titles and abstracts were screened independently by both authors. Full texts of potentially eligible publications were reviewed independently by both authors against inclusion criteria, with discrepancies resolved by discussion. Data were extracted from eligible publications by the lead author, using a modified Cochrane Data Collection Tool and are tabulated in Table 2 (trial characteristics; participant demographics, psychotic symptoms, entry criteria, PD diagnostic criteria, measures used, titration schedule) and Table 3 (descriptive results; psychosis outcomes, motor outcomes, dropouts and adverse events). Raw numerical data for included publications are included in Supplementary Materials.
2.7. Quality Assessment
Risk of Bias was assessed by the lead author using RoB 1 (integrated in RevMan 5.4 software) provided by the Cochrane collaboration [55]. Inclusion of studies was not dependent on outcome of risk of bias assessment. Assessment included reviews of selection bias (random sequence generation and allocation concealment), performance bias, detection bias, attrition bias and reporting bias. Domains were assessed as low, high or unclear risk of bias Results are summarised in Appendix A, Figure A3. Domains were judged following guidance in the Cochrane Handbook for Systematic Reviews of Interventions [56].
2.8. Data Synthesis
Due to the anticipated small number and heterogeneity of trials, a full pooled meta-analysis for all APs was not planned a priori. However, where trials of the same AP were comparable in outcome measures and appropriate data were available, limited inverse-variance random-effects meta-analyses were performed using RevMan 5.4 statistical software, provided by the Cochrane Collaboration [55]. A random-effects model was utilised for pooling of estimates to account for clinical and methodological heterogeneity, including differences in study duration and dosing strategies. Statistical heterogeneity was assessed using the I^2^ statistic. Where pooling was not appropriate due to heterogeneity in outcome measures or insufficient data, findings have been synthesised descriptively and grouped by AP to allow comparison of efficacy, motor effects and tolerability, as well as direction and magnitude of effect. Effect sizes were calculated where adequate data were available.
2.9. Reference Management
All references were managed in Mendeley Cite, and data extraction tables were prepared in Microsoft Word. Database search results were managed in Rayyan [57] for further deduplication and completion of manual screening.
3. Results
A total of 1048 publications were returned from database searches. Following initial deduplication “abstract” and “randomised controlled trial” filters were applied as part of the structured a priori search strategy. 124 papers remained and were manually screened to assess eligibility for inclusion. 113 were excluded (reasons provided in PRISMA flow diagram Appendix A, Figure A1). Full text review of eleven publications was completed. One included patients with Dementia with Lewy Bodies and was disregarded [58]. One lacked a control group and was also disregarded [59] as per pre-defined inclusion criteria. Reference lists of included publications were screened for further relevant papers, yielding one further study that met criteria for inclusion [49]. Ten papers comprising a total of eleven individual clinical studies fulfilled the criteria for inclusion, with a total of 645 patients: 129 from the four quetiapine studies, 137 from the two olanzapine studies, 120 from the two clozapine studies and 259 from the two pimavanserin studies. All included studies utilised single active intervention arm versus placebo; no adjustment for multi-armed designs was required. Where a publication reported two distinct randomised trials, these were extracted and analysed as two separate studies. Four investigated quetiapine [45,46,47,48], two olanzapine [49,50], two clozapine [51,52] and two pimavanserin [53,54]. Cognitive exclusion criteria varied between studies. One excluded patient with dementia [54], although no definition of dementia was provided. Unified Parkinson Disease Rating Scale (UPDRS) Part 1 “intellectual impairment” is however recorded in the results. Two excluded patients with either “significant cognitive impairment” or dementia, if unable to complete testing [48,51] without specifying how these patients were assessed.
Formalised cognitive assessment using Mini-Mental State Examination (MMSE) [60] was undertaken in four papers as part of their exclusion criteria screening. Three used an inclusion cut off score of >20 [49,52,53] and one used a score of >21 [47]. The remaining three papers did not exclude patients on the basis of cognitive impairment or diagnosis of dementia, however, did complete MMSE testing [45,46,50].
Psychosis inclusion criteria differed between the included studies with regards to severity and instruments used to screen participants. One paper [54] utilised criteria developed for diagnosis of PDP, however the remaining nine used standard definitions of psychosis, not specific to PD. Three focused exclusively on hallucinations [47,48,49], with the remainder investigating both hallucinations and delusions.
Diagnostic criteria for Parkinson’s disease was not consistent for the included papers: five citing UK Brain Bank Criteria [45,46,48,52,53]; one utilising Diagnostic and Statistical Manual of Mental Disorders-IV criteria [50]; two papers diagnosed PD on either two or three “cardinal features” with no obvious exclusionary criteria for other parkinsonian presentations [49,51], and two did not report diagnostic criteria [47,54].
Risk of Bias assessment of the included studies found that both blinding of participants and personnel as well as blinding of outcome assessment were all considered to be low risk, however many studies did not describe how allocation concealment was maintained, leading to unclear risk of selection bias in eight of the included papers. Of particular note was the overall high risk of attrition bias in the papers, with no included study considered to be low risk. This may reduce confidence in the interpretation of treatment effects.
Evidence Synthesis
Where pooling of data was undertaken for individual agents, a random-effects model was used to account for anticipated clinical and methodological heterogeneity. Due to the small number of included studies, statistical heterogeneity, as assessed by the I^2^ statistic, was interpreted cautiously.
Of the four quetiapine trials, three found that there was no statistically significant improvement in psychosis of any measure [45,46,47], with one paper showing significant improvements in Brief Psychiatric Rating Scale (BPRS) item II (hallucinations) and Clinical Global Impression-Severity (CGI-S) [48]. No significant worsening of motor symptoms was seen between groups. Meta-analysis was not undertaken for quetiapine due to incomplete data, omitting mean change difference (MD) ± Standard Deviation (SD) or Standard Error (SE).
Both olanzapine studies [49,50], demonstrated no statistically significant improvement in psychosis compared with placebo and showed significant worsening of motor function. Pooled analyses for psychosis and motor symptoms were completed for two olanzapine trials from one paper [50], with statistically significant worsening of motor symptoms observed on UPDRS mean change difference (Figure 1).
Both publications investigating clozapine [51,52] found significant improvements in measures of psychosis with a large effect size, and no statistically significant worsening of motor symptoms compared with placebo. These studies were sufficiently homogeneous in design and outcome measures to allow for pooling of data. Random-effects meta-analysis demonstrated a significant improvement in psychotic symptoms as measured by CGI-S scale mean change difference (Figure 2), with no statistically significant worsening of motor scores as measured by UPDRS (Figure 3).
Statistical heterogeneity within the analyses in this review was low, which may indicate consistent effects between pooled studies; however, this must be interpreted cautiously due to the small number of trials included for each agent.
Pimavanserin studies found no statistically significant worsening of motor symptoms, however differed in improvements in psychosis. One paper did not reach significance in primary efficacy outcome measures but found global rater-assessed measure of hallucinations and delusions to have improved significantly when compared with placebo [54]. One paper found significant improvement in psychosis using both primary and secondary efficacy measures when compared with placebo [53], however failed to reach a previously specified threshold for meaningful clinical relevance. Due to differences in the reporting of data between the two pimavanserin studies (one providing MD ± SE for both psychosis and motor symptoms, and one paper providing baseline and post treatment means ± SD, with no MD ± SD/SE), pooled analysis was not performed.
4. Discussion
Across the included studies, clozapine demonstrated the most consistent efficacy for treatment of PDP showing a large effect size in most measures of psychosis utilised by the papers, with only a single measure marginally missing the large effect threshold value of 0.8. Effect sizes are presented in Appendix A, Table A1, calculated using data provided by the included papers where available. Both studies’ CGI-S measure has shown a robust clinician rated treatment effect for PDP, which supports the significant treatment effect seen in both patient-rated BPRS and Positive and Negative Syndrome Scale (PANSS) positive.
UPDRS motor scores were not significantly different between groups in either paper, however a trend towards improvement was seen in those treated with clozapine. The Parkinson Study Group noted that there was an improvement in tremor in the clozapine group, not reported in Pollak et al., (2014) [52], which may account for this. Pollak et al. [52], report seven AEs of worsening Parkinsonism, however these did not require cessation of medication and were not reflected in overall motor scoring. Pollak et al., [52] also reported a high frequency of somnolence in the active group, which may reflect the relatively rapid titration of treatment when compared with the Parkinson Study Group, (1999) [51] paper. Both clozapine studies experienced at least one instance of neutropenia (NP) or leukopenia, which highlights the potential risk of inducing haematological abnormalities despite the low doses used. These findings broadly support the conclusions from the previous meta-analyses in Table 1, all of which showed consistent improvements in measures of psychosis from their included trials.
The studies using pimavanserin show a mixed picture with regards to efficacy, which is at odds with results from previous meta-analyses possibly due to the exclusion of two unpublished trials that are frequently included, as well as other trials including comparator studies that were not included in this review due to a priori inclusion/exclusion criteria. Both included pimavanserin studies in this review utilise the Scale for the Assessment of Positive Symptoms, Hallucinations and Delusions subsections (SAPS H + D) as a measure of psychosis, however Cummings et al., [53] showed a significant improvement, whilst Meltzer et al. [54], efficacy results fell short of significance. Mean SAPS H + D score reductions for pimavanserin groups in both studies appear similar, as do the mean score reductions in placebo groups. Considering the greater treatment effect seen by Meltzer [54], the lack of significance may be the result of a type 2 error, potentially due to relatively small sample size. Cummings et al., [53] utilised the Scale for the Assessment of Positive Symptoms—Parkinson’s Disease (SAPS-PD) as their primary outcome measure for psychosis, which is unique to this paper. It is unclear if this measure has been robustly validated as a measure of PDP, however the effect size seen is consistent with other measures of psychosis used in the study, which would suggest that this may be a valid measure of PDP. The authors have stated that an improvement of 1 on Clinical Global impression—Improvement (CGI-I) would be deemed to be a clinically meaningful improvement in PDP, which was associated with a change of 2.33 in SAPS-PD during its development [61]. Cummings et al., [53] did not observe an improvement of 1 on CGI-I, the benchmark for meaningful clinical improvement, despite SAPS-PD mean reduction of 3.06. Statistical significance of individual reported rates side effects for both studies have not been reported, however both studies appear to show an increased risk of peripheral oedema. Data from both studies show no statistically significant worsening of motor symptoms, which is consistent with pimavanserin’s very low D_2_ binding affinity (detailed in Table 4) resulting in negligible nigrostriatal DA blockade.
BPRS, CGI-S and UPDRS outcomes for the Breier et al., [50] studies support the findings of Ondo et al., (2002) [49] with olanzapine’s lack of efficacy for the treatment of PDP, as well as its negative impact on motor symptoms of PD. The AEs reported in the Ondo et al., USA study [47,49] add weight to the results, with significantly higher reported rates of extrapyramidal symptoms, hallucinations and increased salivation in the active group, however these differences were not seen in the European study. These results are in keeping with olanzapine’s comparatively high D_2_ receptor binding affinity, with previous studies [62] showing significant D_2_ receptor occupancy at low doses of olanzapine, comparable to mean dosages used in the included studies. This may explain PD patients’ particular sensitivity due to depleted nigrostriatal DA being subject to further blockade, worsening motor and extrapyramidal symptoms.
Three of the four studies utilising quetiapine included in this review showed no significant improvement in psychosis over placebo, however Fernandez et al., (2009) [48] reported significant improvement in two measures, with improvements in BPRS item 12 demonstrating a large effect size. The reasons for these significant results are unclear but may represent a type 1 error due to small sample size and method used for intention-to-treat analysis. All quetiapine studies showed no significant worsening of motor scores, again possibly due to quetiapine’s low D_2_ receptor binding affinity, again broadly supporting the findings of previously published literature.
The studies that have been included in this review suffered several methodological limitations that may affect their reliability. Power calculations were provided in only five of the eleven included studies [45,46,51,53,54], with only two of these recruiting the required numbers of participants [51,54]. A further significant limitation of the literature included in this review was incomplete outcome data, which limited statistical analysis, with only six studies providing adequate data required to calculate effect size appropriately (Appendix A, Table A1). Study design heterogeneity, including duration, dosages of medications, and titration schedules further complicates direct comparison, as does the use of differing scales for the assessment of psychosis. Entry criteria were also not consistent for all studies, with differing inclusion and exclusion thresholds for both cognitive function and psychosis. PDP has no formalised diagnostic criteria, however a National Institute of Neurological Disorders and Stroke/National Institute for Mental Health work group developed suggested criteria [65] which have been utilised by two of the included publications. Other publications in this review either conducted their studies prior to the publication or have used an alternative definition of PDP. This introduces further participant heterogeneity between the papers. Five studies were deemed to have an unclear risk of attrition bias, four of which utilised Last Observation Carried Forward for missing data. This method assumes stability of symptoms after dropout, and may introduce bias if attrition differs between groups. A further six studies were deemed high risk of attrition bias (see Appendix A, Figure A3). Incomplete follow up and potential bias reduce confidence in the reliability of calculated treatment effect estimates across studies.
The findings of this review are broadly consistent with the current body of evidence for the AP management of PDP (see Table 1), with clozapine demonstrating the most consistent efficacy signal and olanzapine showing poor efficacy alongside a propensity to worsen motor scores. Most recent reviews similarly find that although quetiapine does not appear to exacerbate motor symptoms in PD patients, its antipsychotic effect is comparable to placebo. The quetiapine trial demonstrating statistically significant improvements in CGI-S and the hallucinations subsection of BPRS (item 12) was associated with a 50% attrition rate in the active intervention arm, with non-completer data imputed using a linear model of completers. High attrition and small sample size reduces confidence in the robustness and reliability of these findings [48]. Pimavanserin is widely reported to produce statistically significant improvements in psychosis scores and remains a potential alternative to clozapine; however, the effect sizes observed for the included studies in this review were small to medium, and clinically meaningful improvements were not achieved in one of the two trials. This highlights that statistical significance within trial data does not necessarily translate into clinically meaningful benefit for patients.
5. Conclusions
Recent publications examining AP prescribing patterns indicate quetiapine accounts for approximately half of all prescriptions for the treatment of PDP. National Institute for Health and Care Excellence (NICE) guidelines recommend considering quetiapine in patients without cognitive impairment and offering clozapine where standard management is ineffective [28]. The findings from this systematic review suggest that, despite quetiapine’s low risk of worsening motor symptoms in PD, it has not demonstrated benefit over placebo for the treatment PDP in double-blind, randomised, placebo-controlled trials. Given the increased risk of mortality in patients prescribed APs, a clear therapeutic benefit should be seen to justify their use.
Currently available evidence does not support the use of quetiapine as a first-line AP for the treatment of PDP, particularly when other agents have demonstrated superiority to placebo, without appreciable worsening of motor scores. Notably, although efficacious for PDP, clozapine continues to carry a risk of significant adverse effects including NP even with the low dosages utilised in PDP trials. Owing to the potential for significant and life-threatening adverse effects, stringent haematological monitoring has historically been required for patients receiving clozapine. Perceived risk and monitoring burden may impact clinicians’ willingness to prescribe clozapine for PDP, potentially contributing to its relatively low utilisation despite evidence of efficacy and overall tolerability. This may in turn result in some patients’ psychosis remaining inadequately managed.
More recently, clozapine monitoring guidance in the European Union has changed to 12-weekly blood tests from the previous four-weekly if the patient has not developed NP within the first year of treatment, and then moving to yearly testing if no evidence of NP in the first two years. This is based upon large meta-analyses [66], retrospective cohort studies [67] and registry-based analyses [68,69], which have found NP to be most common in early treatment, with incidence decreasing after 18 weeks. These evolving guidelines may reduce the burden of haematological monitoring and may improve the utilisation of clozapine for the treatment of PDP due to changing perspectives on risk, and improved capacity in dedicated clozapine clinics from these reduced monitoring requirements.
A collaborative, multidisciplinary approach involving both movement disorder and mental health specialists is advised when managing PDP, due to its impact on quality of life, morbidity and mortality. Dedicated PDP clozapine services and early involvement of community mental health teams to robustly monitor for PDP may improve long-term outcomes for PD patients.
The strengths of this review include its scope; completing a systematic search for publications investigating the efficacy and tolerability of APs in the treatment of PDP over four scientific databases, including all currently available APs worldwide to reduce the likelihood of arbitrarily disregarding studies completed with APs not available in the UK. Only primary data has been utilised; double-blind, randomised, placebo-controlled studies that have been published in a peer reviewed journal have been included, providing the highest level of evidence for treatment effect.
The limitations of this review include: its restriction of eligible studies to published, peer reviewed double-blind, randomised, placebo-controlled trials, which excluded double-blind comparator studies that may have provided comparative effectiveness data for a wider range of APs. This limits the number of trials and agents included in this review, which may impact its clinical utility. Limiting papers to English language may have also resulted in relevant papers being omitted. Use of automated database search limits may arbitrarily disregard relevant studies, and a lack of preregistration limits the verifiability of a priori review protocol. However, narrowly defined inclusion criteria, restricted to published, peer reviewed double-blind placebo-controlled trials mitigates the risk of selective inclusion exclusion of relevant studies. The exclusion of unpublished trials which may be listed on clinical trial registries may also contribute to publication bias, potentially resulting in data synthesis supporting favourable efficacy or tolerability.
Despite the high prevalence of PDP, there is a paucity of high-quality double-blind, randomised placebo-controlled clinical trials that inform national guidelines, with only four AP medications investigated to this standard of evidence. Among these, inter-study design heterogeneity, as well as limited number of included studies render statistical analysis of provided data more difficult to interpret. Further clinical trials of AP medications, particularly those with low D_2_ binding affinity, may help to improve the evidence base that is currently used to inform national prescribing guidelines. Direct head-to-head pimavanserin clozapine studies may more clearly demonstrate comparative efficacy and tolerability, as well as comparator studies between pimavanserin and quetiapine—the most commonly prescribed AP medication for PDP in the UK [29,32]. Tolerability and long-term safety studies for newer agents such as pimavanserin, as well as the currently NICE recommended clozapine in PD cohorts would be of significant benefit for clinicians to better understand balance of risk when prescribing for their patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Giguère N. Nanni S.B. Trudeau L.E. On cell loss and selective vulnerability of neuronal populations in Parkinson’s disease Front. Neurol.2018945510.3389/fneur.2018.0045529971039 PMC 6018545 · doi ↗ · pubmed ↗
- 2Dorsey E.R. Elbaz A. Nichols E. Abbasi N. Abd-Allah F. Abdelalim A. Adsuar J.C. Ansha M.G. Brayne C. Choi J.-Y.J. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016 Lancet Neurol.20181793995310.1016/S 1474-4422(18)30295-330287051 PMC 6191528 · doi ↗ · pubmed ↗
- 3GBD 2015 Neurological Disorders Collaborator Group Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015 Lancet Neurol.20171687789710.1016/S 1474-4422(17)30299-528931491 PMC 5641502 · doi ↗ · pubmed ↗
- 4Okunoye O. Marston L. Walters K. Schrag A. Change in the incidence of Parkinson’s disease in a large UK primary care database NPJ Park. Dis.202282310.1038/s 41531-022-00284-0PMC 892419435292689 · doi ↗ · pubmed ↗
- 5Postuma R.B. Berg D. Advances in markers of prodromal Parkinson disease Nat. Rev. Neurol.20161262263410.1038/nrneurol.2016.15227786242 · doi ↗ · pubmed ↗
- 6Pellicano C. Dario B. Pisani V. Buttarelli F. Giovannelli M. Pontieri F.E. Prodromal non-motor symptoms of Parkinson’s disease Neuropsychiatr. Dis. Treat.2007314515110.2147/nedt.2007.3.1.14519300544 PMC 2654529 · doi ↗ · pubmed ↗
- 7Poewe W. Non-motor symptoms in Parkinson’s disease Eur. J. Neurol.200815142010.1111/j.1468-1331.2008.02056.x 18353132 · doi ↗ · pubmed ↗
- 8Marsili L. Rizzo G. Colosimo C. Diagnostic criteria for Parkinson’s disease: From James Parkinson to the concept of prodromal disease Front. Neurol.2018915610.3389/fneur.2018.0015629628907 PMC 5877503 · doi ↗ · pubmed ↗