Precision Diagnosis in APOL1 Kidney Disease With the p.N264K M1 Protective Variant
Elena Martinelli, Juntao Ke, Atlas Khan, Janewit Wongboonsin, David R. Vanderwall, Tze Y. Lim, Dominick Santoriello, Yask Gupta, Michelle T. McNulty, Satoshi Koyama, Sidhant Puntambekar, Andrew S. Bomback, Pietro Canetta, Matthias Kretzler, Giovanni Montini, William Morello

TL;DR
This study shows that the M1 variant in APOL1 can help distinguish APOL1-related kidney disease from other types, suggesting a potentially treatable cause.
Contribution
The study demonstrates that the M1 variant acts as a genetic modifier in APOL1-related kidney disease, improving diagnostic precision.
Findings
The M1 variant is inversely associated with FSGS/SRNS in individuals with APOL1 high-risk genotypes.
APOL1-HR individuals with M1 are four times more likely to have non-APOL1 CKD.
M1 presence in APOL1-HR CKD cases is linked to alternative, treatable causes identified via medical records and biopsies.
Abstract
This case-control study evaluates whether, in patients with APOL1 high-risk genotype kidney disease with at least 1 G2 allele, M1 can distinguish APOL1 chronic kidney disease from non-APOL1 chronic kidney disease. What is the association of the protective APOL1 M1 (p.N264K) variant with kidney disease risk in individuals with APOL1 high-risk (APOL1-HR) and low-risk (APOL1-LR) genotypes? In this case-control study of 107 696 individuals, the M1 variant was associated with improved diagnostic precision for kidney disease causes among individuals with APOL1-HR genotypes and showed no independent association with protection in the general population of individuals with APOL1-LR genotypes. These findings suggest that presence of the M1 variant in individuals with APOL1-HR genotypes may point to a distinct and potentially actionable mechanism of kidney disease. The APOL1 M1 (p.N264K)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
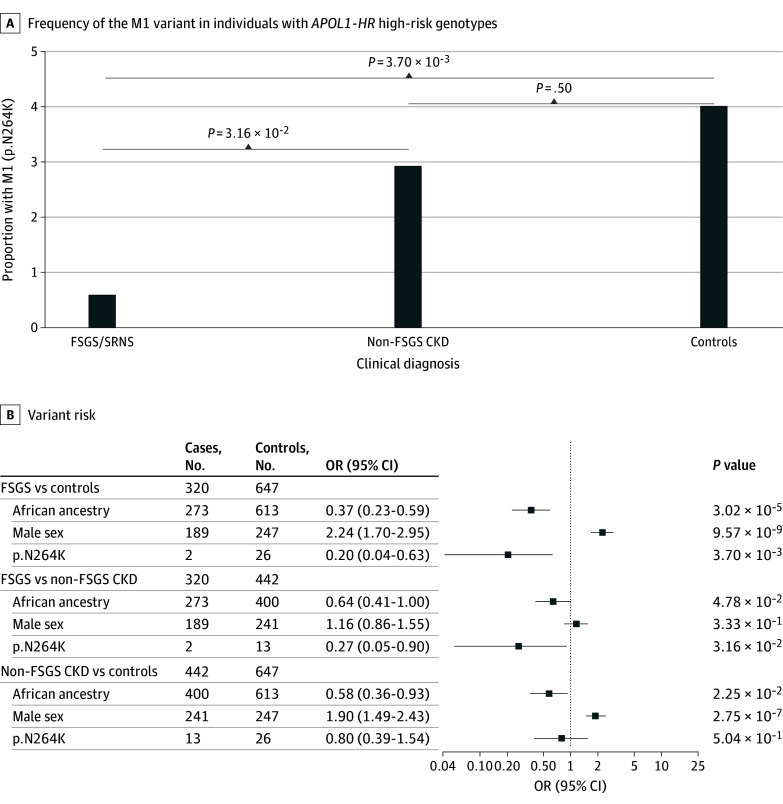 Figure 1
Figure 1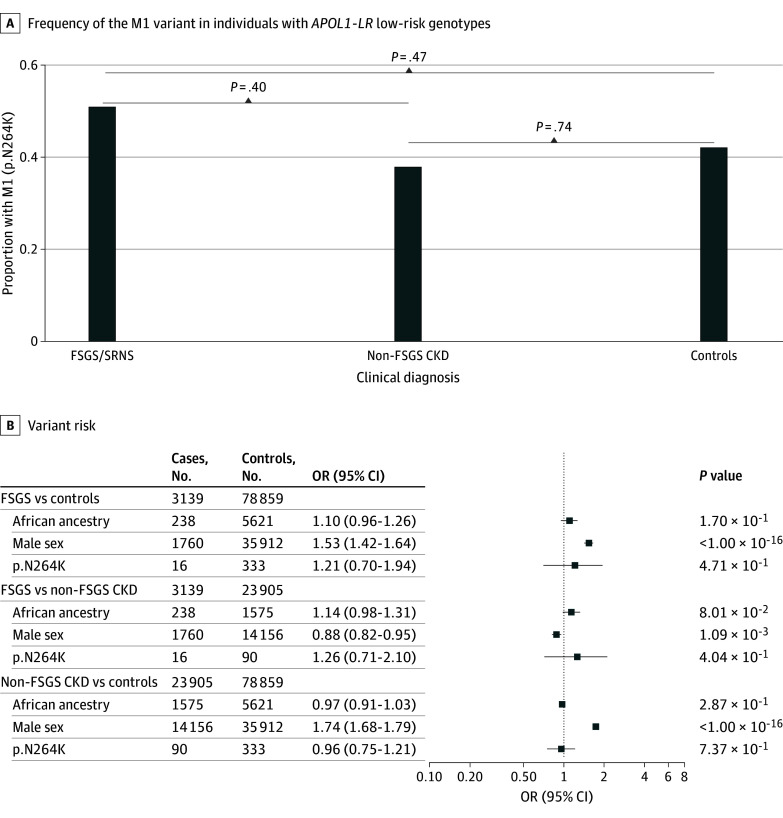 Figure 2
Figure 2| Characteristic | FSGS or SRNS (n = 3460) | Non-FSGS CKD (n = 24 382) | Control (n = 79 854) |
|---|---|---|---|
| Sex | |||
| Female | 1510 (44.0) | 9961 (41.0) | 43 523 (55.0) |
| Male | 1950 (56.0) | 14 421 (59.0) | 36 270 (45.0) |
| Unknown | 0 | 0 | 61 (<0.1) |
| Ancestry | |||
| African | 511 (14.8) | 1975 (8.1) | 6293 (7.9) |
| East Asian | 71 (2.1) | 558 (2.3) | 1571 (2.0) |
| European | 1759 (50.8) | 17 775 (72.9) | 58 941 (73.8) |
| Multiethnic | 971 (28.1) | 3464 (14.2) | 11 694 (14.6) |
| South Asian | 147 (4.2) | 575 (2.4) | 1068 (1.3) |
| Unknown | 1 (<0.1) | 35 (0.1) | 287 (0.4) |
| Low | 3140 (91.0) | 23 940 (98.0) | 79 203 (99.0) |
| G0G0 | 2948 (85.0) | 22 982 (94.0) | 75 778 (95.0) |
| G0G1 | 120 (3.5) | 574 (2.4) | 1965 (2.5) |
| G0G2 | 72 (2.1) | 384 (1.6) | 1460 (1.8) |
| High | 320 (9.2) | 442 (1.8) | 651 (0.8) |
| G1G1 | 158 (4.6) | 191 (0.8) | 248 (0.3) |
| G1G2 | 125 (3.6) | 196 (0.8) | 306 (0.4) |
| G2G2 | 37 (1.1) | 55 (0.2) | 97 (0.1) |
| Wild-type | 3442 (99.0) | 24 278 (100) | 79 493 (100) |
| Heterozygous | 18 (0.5) | 103 (0.4) | 352 (0.4) |
| Homozygous | 0 | 1 (<0.1) | 9 (<0.1) |
| Alleles and genotypes | African | East Asian | European | Multiethnic | South Asian | FSGS | CKD | Controls |
|---|---|---|---|---|---|---|---|---|
| Alleles | ||||||||
| G0 | 11 502 (65.5) | 4396 (99.9) | 3572 (99.8) | 31 017 (96.2) | 3572 (99.8) | 6088 (88.0) | 46 922 (96.2) | 154 981 (97.0) |
| G1 | 3758 (21.4) | 4 (0.1) | 41 (<0.1) | 657 (2.0) | 5 (0.1) | 561 (8.1) | 1152 (2.4) | 2767 (1.7) |
| G2 | 2298 (13.1) | 0 | 25 (<0.1) | 584 (1.8) | 3 (0.1) | 271 (3.9) | 690 (1.4) | 1960 (1.2) |
| Genotypes | ||||||||
| G0G0 | 4009 (45.7) | 2197 (99.9) | 78 410 (99.9) | 15 009 (93.1) | 1782 (99.6) | 2948 (85.2) | 22 982 (94.3) | 75 778 (94.9) |
| G0G1 | 2092 (23.8) | 2 (0.1) | 39 (0.1) | 509 (3.2) | 5 (0.3) | 120 (3.5) | 574 (2.4) | 1965 (2.5) |
| G0G2 | 1392 (15.9) | 0 | 25 (<0.1) | 490 (3.0) | 3 (0.2) | 72 (2.1) | 384 (1.6) | 1460 (1.8) |
| G1G1 | 547 (6.2) | 1 (0.1) | 1 (<0.1) | 48 (0.3) | 0 | 158 (4.6) | 191 (0.8) | 248 (0.3) |
| G1G2 | 572 (6.5) | 0 | 0 | 52 (0.3) | 0 | 125 (3.6) | 196 (0.5) | 306 (0.4) |
| G2G2 | 167 (1.9) | 0 | 0 | 21 (0.1) | 0 | 37 (1.1) | 55 (0.2) | 97 (0.1) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal Diseases and Glomerulopathies · Coagulation, Bradykinin, Polyphosphates, and Angioedema · Complement system in diseases
Introduction
Two West African ancestry–associated variants in the apolipoprotein L1 (APOL1) gene, G1 (rs73885319 and rs60910145) and G2 (rs71785313), contribute to the 5-times higher incidence of chronic kidney disease (CKD) among Black Americans compared with European Americans, who preferentially carry the G0 wild-type (WT) haplotype.^1^
A spectrum of nondiabetic kidney diseases is associated with APOL1 high-risk (HR) genotypes, defined as G1G1, G1G2, and G2G2. Among these conditions, APOL1-HR genotypes confer large odds ratios (ORs) for focal segmental glomerulosclerosis (FSGS), a pattern of glomerular injury underlying a large fraction of idiopathic nephrotic syndrome, when compared with individuals with APOL1 low-risk genotypes (APOL1-LR), G0G0, G0G1, G0G2.^2,3,4^
However, only 5% to 8% of individuals with *APOL1-*HR genotypes actually develop FSGS, suggesting that environmental and/or genetic factors modify their penetrance.^5^ For genetic testing to be informative in both ruling in or out a diagnosis and, hence, providing enhanced and equitable care, the genotype of interest must display a high deterministic value on the clinical presentation of the tested individual. This is especially true in genetics-first or genetics-early settings, when genetic testing is administered early in the diagnostic workup and its results might influence physicians’ decision-making regarding further diagnostic testing (eg, imaging studies and kidney biopsy) or treatment (eg, avoidance of immunosuppression and starting targeted therapies). APOL1-HR genotypes, despite their large effect size for risk of FSGS (ORs up to 30)^2^ and CKD (ORs up to 4),^6^ have high prevalence but low penetrance in individuals of genetic African ancestry, consequently complicating the diagnostic, prognostic, and therapeutic workup for APOL1 kidney disease. On one hand, early knowledge of APOL1 risk variant status during the diagnostic workup can improve diagnostic precision, avoid unnecessary treatments, and identify individuals who might benefit from specific APOL1 targeting approaches.^7,8^ On the other hand, APOL1-HR genotypes are present in approximately 13% of self-reported Black US residents^9,10^ and in up to 50% of individuals living in West Africa^11,12,13^; this high frequency, together with their incomplete penetrance and limited predictive value, poses risks for so-called label diagnosis that might result in undesired effects, such as incomplete diagnostic work-up (eg, avoidance of kidney biopsy) or modifying standard-of-care management decisions with the unwanted possibility of increasing disparities.
We, among others, reported on the protective association between the APOL1 p.N264K missense variant (M1; rs73885316) and APOL1 kidney disease and, specifically, FSGS.^14,15^ The APOL1 M1, when coinherited on the same haplotype with the high-risk G2 APOL1 allele,^16^ virtually negates this association, effectively rendering G2-containing APOL1 HR genotypes comparable with nonrisk genotypes.^14,15,17,18^
These new insights support the potential importance of genotyping the APOL1 M1 variant in routine genetic testing.^19,20,21^ In the setting of presymptomatic screening or transplant donor evaluation, the presence of M1 in individuals with APOL1 G1G2 and G2G2 genotypes—traditionally considered to be at high risk—would lower the estimated risk of them developing CKD and FSGS. In the setting of patients with already diagnosed steroid-resistant nephrotic syndrome (SRNS) or FSGS, CKD, or end-stage kidney disease, finding APOL1 G1G2 or G2G2 and M1 on genetic testing would strongly suggest that they did not have APOL1 kidney disease. Rather, these patients would require a different and complete diagnostic workup to identify immune, toxic, structural, or other causes for their disease and enact adequate therapies. These observations led to our first hypothesis: in individuals with APOL1-HR genotypes, M1 genotyping will improve the diagnostic precision for APOL1 kidney disease and will help distinguishing it from non-APOL1 CKD or FSGS.
Additionally, in the original study by Hung et al,^15^ the authors observed a suggestive protective association of M1 even in individuals with APOL1-LR genotypes. If confirmed, this protective association of M1 in APOL1-LR genotype carriers would imply a potentially deleterious effect of APOL1 even in individuals with G0, which has not been previously demonstrated. It would additionally suggest an independent absolute protective role of M1 against CKD development. Current understanding of APOL1-associated kidney toxicity contradicts a toxic role of G0.^22,23,24^ Therefore, our study aimed to investigate a second hypothesis: in individuals with APOL1-LR genotypes, M1 may have a protective association against FSGS and CKD.
To test these hypotheses, we studied 107 696 individuals from 2 large tertiary hospitals’ biobanks at Columbia University Irving Medical Center (CUIMC) and the Massachusetts General Brigham Health system. We then validated the results in 23 955 individuals of African ancestry from the Electronic Medical Records (EMR) and Genomics (eMERGE-III) project, the UK Biobank (UKB), and the All of Us (AoU) research program.
Methods
For this case-control study, written informed consent was collected from all participating individuals and/or their guardians in accordance with the Columbia University institutional review board and the policy on bioethics and human biologic samples of AstraZeneca. Results conform to Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline. Additional details on study methods are available in the eMethods in Supplement 1.
Cohorts and DNA Sequencing
The total discovery cohort included 54 304 individuals from CUIMC^25^ and 53 392 individuals from the Mass General Brigham Biobank^26^ with genome or exome sequencing (eTable 1 in Supplement 1). This dataset included 27 842 individuals with CKD and 79 854 individuals not meeting the CKD criteria who were defined as controls. Among individuals with CKD, 3460 had biopsy-confirmed FSGS or SRNS and 24 382 had CKD not attributable to FSGS or SRNS (here defined as non-FSGS CKD). A total of 79 854 controls were identified from individuals without evidence of CKD. KING version 2.3.1 (KING)^27^ was used to remove related individuals; principal components were calculated using PLINK2^28^ to infer genetic ancestries according to the 1000 Genome populations,^29^ and APOL1 G1, G2, and M1 genotypes were extracted from sequencing data (eFigure 1 in Supplement 1).
Replication in Public Biobanks
We replicated our findings in individuals of African ancestry across 3 biobanks: the EMR and eMERGE-III project,^30,31^ the UKB,^32^ and the AoU research program.^33,34,35^ Ancestry was defined using a random forest-based machine learning approach. We trained and tested the random forest model using subjects from the 1000 Genomes Project with known ancestry labels.^29^
To define individuals with CKD and controls in these 3 biobanks, we applied our validated CKD e-phenotyping algorithm.^31^ To classify individuals with CKD caused by FSGS or SRNS, we developed a selection pipeline based on relevant International Classification of Diseases, Ninth Revision (ICD-9) codes (eTable 2 and eFigure 2 in Supplement 1), generating 3 cohorts aligned with the discovery study: FSGS or SRNS, non-FSGS CKD, and controls.
Statistical Analyses
In the discovery cohorts, pairwise comparisons of M1 variant prevalence across phenotypic categories (FSGS or SRNS, non-FSGS CKD, and controls) were performed using Fisher exact test and regression analyses were performed using the Firth bias-reduced logistic regression, which applies a penalized likelihood approach to mitigate small-sample bias and issues of separation.^36,37^ Covariates included sex and genetic ancestry in the full-cohort models. Conversely, models restricted to individuals of African ancestry were adjusted for sex only. In the external biobank datasets, M1 variant prevalence was compared across phenotypic groups using the Cochran-Mantel-Haenszel test, with biobank and sex as stratifying variables. Statistical significance was defined as a 2-sided P value less than .05. All analyses and forest plots were conducted with R version 4.5.1 (R Project for Statistical Computing) and the bar plots were generated using GraphPad Prism version 10.6.1 for Windows (GraphPad Software).
Results
Baseline Characteristics of the Discovery Cohort and APOL1 Variants
The combined cohort included 107 696 individuals (54 994 [51.1%] female; 8779 [8.2%] African ancestry, 78 475 [72.9%] European ancestry, and 16 129 [15.0%] multiethnic ancestry), including 3460 with FSGS, 24 382 with non-FSGS CKD, and 79 854 controls (Table 1). Among those with FSGS, 1510 of 3460 were female (43.6%); sex distributions were similar in non-FSGS CKD (9961 of 24 382 [40.8%]), while controls had a slightly higher proportion of female participants (43 523 of 79 854 [54.5%]).
Genetic ancestry distributions reflected the diversity of the dataset. Of 3460 individuals with FSGS, 511 (14.8%) were of African ancestry, 71 (2.1%) of East Asian ancestry, 1759 (50.8%) of European ancestry, 971 (28.1%) of multiethnic ancestry, and 147 (4.2%) of South Asian ancestry. Of 24 382 individuals in the non-FSGS CKD group, 1975 (8.1%) were of African ancestry, 558 (2.3%) were of East Asian ancestry, 17 775 (72.9%) were of European ancestry, 3464 (14.2%) were of multiethnic ancestry, and 575 (2.4%) were of South Asian ancestry. Of 79 854 in the control group, 6293 (7.9%) were of African ancestry, 1671 (2.0%) were of East Asian ancestry, 58 941 (73.8%) were of European ancestry, 11 694 (14.6%) were of multiethnic ancestry, and 1068 (1.3%) were of South Asian ancestry.
APOL1 risk genotypes followed expected patterns across disease groups. APOL1-HR genotypes were observed in 320 of 3460 individuals (9.2%) of all-ancestries FSGS (273 of 320 [85.3%] of African ancestry, 46 of 320 [14.4%] of multiethnic ancestry, and 1 [0.3%] of European ancestry), 442 of 24 382 individuals (1.8%) with CKD, (400 of 442 [90.5%] of African ancestry, 41 of 442 [9.3%] of multiethnic ancestry, and 1 of 442 [0.2%] of East Asian ancestry), and 651 of 19 854 (0.8%) controls (613 of 651 [94.2%] of African ancestry, 34 of 651 [5.2%] of multiethnic ancestry and 4 of 651 [0.6%] of unknown ancestry), with the remainder in each group classified as APOL1-LR.
As expected, individuals of African ancestry had the highest allelic frequencies for G1 and G2 as compared with all the other groups (Table 2). APOL1 M1 allelic frequency and distribution among groups are summarized in eTable 3 in Supplement 1. A total of 61 controls did not have sex data (all APOL1-LR), and for 287 controls (4 APOL1-HR), 35 non-FSGS CKD (all APOL1-LR) cases, and 1 FSGS (APOL1-LR) case, a genetic ancestry category could not be estimated; these 384 cases were removed from the Firth bias-reduced logistic regression.
APOL1 M1 Variant Association With FSGS or SRNS
To investigate our first hypothesis (ie, that in individuals with APOL1-HR, M1 genotyping will improve the precision of diagnosing APOL1 kidney disease and distinguishing it from non-APOL1 kidney disease), we investigated the association of the M1 variant in the APOL1-HR group (G1G1, G1G2, and G2G2) with CKD cases and controls. M1 was nominally inversely associated with CKD status overall and when compared with controls (15 of 762 patients [1.97%] vs 26 of 651 [3.99%]; Fisher exact OR, 0.48; 95% CI, 0.24-0.96; P = 2.62 × 10^−2^; Firth regression OR, 0.54; 95% CI, 0.28-1.02; P = 5.83 × 10^−2^). This inverse association between M1 and cases was primarily associated with the FSGS or SRNS subgroup (2 of 320 patients [0.63%]) compared with controls (26 of 651 patients [3.99%]; Fisher exact OR, 0.15; 95% CI, 0.02-0.61; P = 1.86 × 10^−3^; 26 of 647 patients; Firth regression OR, 0.20; 95% CI, 0.04-0.63; P = 3.70 × 10^−3^) (Figure 1; eTable 4 in Supplement 1). However, M1 was not significantly associated with non-FSGS CKD cases (13 of 442 patients [2.94%]) or controls (Fisher exact OR, 0.73; 95% CI, 0.34-1.49; P = .41; 13 of 442 patients; Firth regression OR, 0.80, 95% CI, 0.39-1.54; P = .50) (Figure 1; eTable 4 in Supplement 1). Furthermore, harboring M1 was approximately 4 times more likely to be associated with non-FSGS CKD than FSGS or SRNS cases (13 of 442 patients [2.94%] vs 2 of 320 [0.63%], Fisher exact OR, 4.81; 95% CI, 1.08-44.21; P = 3.15 × 10^−2^; Firth regression OR, 3.70; 95% CI, 1.11-18.96; P = 3.16 × 10^−2^) (Figure 1; eTable 4 in Supplement 1).
M1 Association With Focal Segmental Glomerulosclerosis (FSGS) or Steroid-Resistant Nephrotic Syndrome (SRNS), Non-FSGS Chronic Kidney Disease (CKD), and Controls in APOL1 High-Risk (HR) GenotypesThe bar plots show the frequency of the M1 variant (p.N264K) among individuals with APOL1-HR genotypes, stratified by clinical diagnosis: FSGS or SRNS, non-FSGS CKD, and unaffected controls. Forest plots show odds ratios (ORs) and 95% CIs from the Firth bias-reduced logistic regression analysis comparing individuals with FSGS or SRNS with controls and with non-FSGS CKD, as well as non-FSGS CKD with controls. Covariates included genetic ancestry (African and non-African; non-African as reference), sex (female as reference) and M1 (wild-type as reference). LR indicates low-risk; OR, odds ratio.
Restricting these analyses to individuals with APOL1 G2-containing HR genotypes (G1G2 and G2G2) supported these findings (eFigure 3 in Supplement 1). Moreover, since G1, G2, and M1 are significantly more frequent in individuals of African continental genetic ancestry, we conducted sensitivity analyses in the 673 CKD cases and 613 controls of genetic African ancestry only. This showed nearly identical results as in the whole dataset of individuals with APOL1-HR genotypes (eTable 5 in Supplement 1).
We next proceeded to validate these results in 3 large biobanks. Both individually in eMERGE-III, UKB, and AoU, as well as in aggregate, these analyses showed a direction-consistent effect size between M1 and FSGS or SRNS cases as compared with controls and individuals with non-FSGS CKD, although the results were not statistically significant, possibly due to the limited number of FSGS or SRNS cases in these population-based biobanks (Cochran-Mantel-Haenszel stratified by Biobank and sex, FSGS vs controls common OR, 0; 95% CI, 0.00-1.26; P = .11; FSGS vs non-FSGS CKD common OR, 0; 95% CI, 0.00-1.73; P = .26) (eTable 6, eFigure 4 in Supplement 1).
Altogether, these data confirm that in individuals with APOL1-HR genotypes, the presence of M1 is associated with nearly complete protection against APOL1 FSGS or SRNS. Furthermore, in non-FSGS CKD cases with APOL1-HR genotypes, the presence of M1 suggests a non-APOL1 cause and that an alternative, potentially treatable cause of their disease should be pursued.
Electronic Health Record (EHR) Review and Non-APOL1 Causes for CKD in APOL1-HR M1 Carriers
The large-scale human genetics studies reported previously indicate that the presence of M1 protects against APOL1 kidney disease, especially FSGS or SRNS. Consequently, individuals with CKD who have APOL1-HR and the M1 protective missense variant should have an alternative cause for their CKD.
To investigate this, we conducted a detailed retrospective review of the EHR for all APOL1-HR cases with CKD who harbored the M1 variant in the discovery cohort. A detailed description of the findings is available in eAppendix, eFigure 5, and eTable 12 in Supplement 1. In summary, in the FSGS or SRNS group, the 2 APOL1-HR-M1 cases (0.63%) had presentations that suggest that the FSGS or SRNS phenotype in these 2 cases is associated with non-APOL1 mechanisms, and may simply represent the background prevalence of FSGS or SRNS in cases with APOL1-HR-M1; in the non-FSGS CKD group with APOL1-HR genotypes and M1, we were able to identify an alternative, non-APOL1 mediated cause of CKD in 10 of 13 individuals (76.9%). Importantly, in the 3 cases in whom we could not identify an alternative cause of CKD, the diagnostic workup was incomplete.
Protective Association of M1 Independent of APOL1
We next formally tested the hypothesis that M1 might protect against CKD even in those without an APOL1 HR genotype. In individuals with APOL1-LR genotypes (G0G0, G0G1, and G0G2), the prevalence of M1 was similar and not statistically different across groups (Figure 2; eTables 7-8 in Supplement 1). These data suggested no protective association of M1, independent of APOL1-HR genotypes, for FSGS or SRNS and CKD. The results were not different when restricting the analysis to individuals with APOL1-LR genotypes of genetic African ancestry alone (eTable 9 in Supplement 1).
M1 Association With Focal Segmental Glomerulosclerosis or Steroid-Resistant Nephrotic Syndrome (FSGS or SRNS), Non-FSGS Chronic Kidney Disease (CKD), and Controls in APOL1 Low-Risk (LR) GenotypesThe bar plots show the frequency of the M1 variant (p.N264K) among individuals with APOL1 LR genotypes, stratified by clinical diagnosis: FSGS or SRNS, non-FSGS CKD, and unaffected controls. Forest plots show ORs and 95% CIs from the Firth bias-reduced logistic regression analysis comparing individuals with FSGS or SRNS with controls and with non-FSGS CKD, as well as non-FSGS CKD with controls. Covariates included genetic ancestry (African and non-African; non-African as reference), sex (female as reference) and M1 (wild-type as reference). HR indicates high risk; OR, odds ratio.
Nevertheless, there is mounting evidence of a potential heterozygous risk for G1 and G2 that might have confounded these analyses.^11^ Therefore, we conducted a subset analysis restricted to only individuals with G0G0 genotypes across all ancestries. Here, we also found no evidence of an independent protective association of M1 (eTable 10 in Supplement 1).
We again proceeded to validate these results in 3 large biobanks. Both individually in eMERGE-III, UKB, and AoU, as well as in aggregate, these analyses showed no support for a protective association of M1, independent of APOL1-HR genotypes (eTable 11, eFigure 4 in Supplement 1).
Discussion
The identification of the APOL1 M1 (p.N264K) modifier variant in a patient treated in Ghana for Trypanosoma infection in 2013,^18^ followed by the studies in 2019 and 2023,^15,38^ marked an important phase in the efforts devoted to identifying genetic modifiers of APOL1 kidney disease. Now that M1 has been discovered and validated, the next phase of this effort is understanding the extent and magnitude in which the knowledge of a person’s M1 status can improve the precision of our care for them to provide better prognosis and risk stratification, as well as to identify individuals who will benefit the most from drugs directly and indirectly targeting APOL1.^7,8^
In this case-control study, we investigated the association of the M1 variant across different APOL1 kidney risk genotypes with distinct phenotypic groups within our large-scale, well-characterized study cohort followed by validation in 3 external biobanks. Specifically, we first investigated whether M1 could enhance diagnostic precision for people with APOL1-HR genotypes (prevalently of African ancestry and multiethnic genetic ancestries) who are diagnosed with kidney disease. Second, we tested if M1 could operate as an independent protective modifier against kidney disease in the general APOL1-LR population (ie, in individuals across non-African and African ancestries) in absence of APOL1-HR genotypes.
Therefore, we first assessed if M1 would help in correctly diagnosing APOL1 kidney disease by identifying individuals who, despite harboring recessive APOL1-HR genotypes, had their CKD attributable to non-APOL1 causes, which would require specific diagnostic workup and might be amenable to targeted therapies. As expected, M1 was significantly depleted in FSGS or SRNS APOL1-HR cases compared with non-CKD controls. Interestingly, APOL1-HR cases with non-FSGS CKD only had a marginally lower frequency of M1 as compared with non-CKD controls, but this difference was not statistically significant, and they were approximately 4-fold more likely to harbor M1 as compared with FSGS or SRNS cases. These data suggested that a fraction of APOL1-HR individuals with non-FSGS CKD and who carried the APOL1 M1 variant did not have APOL1 kidney disease but another cause of CKD that would require specific diagnostic workup and treatment. In fact, detailed retrospective EHR review identified a likely alternative non-APOL1 cause of disease for 2 of 2 FSGS and 10 of 13 non-FSGS CKD cases that were APOL1-HR-M1. In particular, the cases spanned across different causes of CKD with a clear independent cause, including diabetic nephropathy, amyloidosis, systemic lupus erythematosus, obstructive uropathy, IgA nephropathy, ANCA-associated vasculitis, immunocomplex mesangial proliferative and sclerosing glomerulonephritis, and steroid-sensitive nephrotic syndrome. Kidney biopsy slides were available for in-depth review in 2 of 13 cases, and both of them had an alternative cause of glomerular disease not attributable to APOL1. Only 2 cases had a clinical diagnosis of hypertension-mediated kidney disease, of which 1 had nonnephrotic range proteinuria that both might be attributable to APOL1, although the possibility of a label diagnosis without a complete diagnostic workup is possible.
Interestingly, even in the FSGS or SRNS group, the 2 cases that were found to harbor a APOL1 high-risk genotype together with the M1 variant had an unlikely APOL1-mediated cause for FSGS, as they were either diagnosed with congenital nephrotic syndrome or had pediatric onset (aged 8 years), nonprogressive FSGS (eAppendix in Supplement 1). In fact, APOL1-HR genotypes are known to increase risk for kidney disease in young adults, but to a lesser extent in children.^39^ Previous studies including pediatric populations showed that the median age of disease onset was older in children who were APOL1-HR compared with children who were APOL1-LR (median [IQR], 11.5 [9.5-12.5] vs 4.5 [1.5-12.5] years), whereas the occurrence of congenital nephrotic syndrome in APOL1-HR individuals remains anecdotical and possibly coincidental.^40^ These data, if further validated, suggest that M1 completely protects against APOL1 FSGS or SRNS, such that in individuals with APOL1-HR-M1 genotypes and FSGS or SRNS, the latter is likely a coincidental diagnosis in the context of primary or secondary podocytopathy which would require a further diagnostic pursuit (ie, testing for Mendelian disease or antinephrin antibodies). Similarly, in individuals with non-FSGS CKD, the presence of M1 suggests an alternative, non-APOL1, cause of kidney disease that would require complete serologic, histologic and radiologic diagnostic workup. While this recommendation is particularly important in the presence of M1, physicians should nevertheless apply standard-of-care diagnostics, including kidney biopsy, to all individuals with APOL1-HR, as M1 does not explain the full incomplete penetrance of APOL1 risk variants, and many of these individuals have alternative etiologies for their CKD. Now that a new *ICD-10 *code, N07.B, has been introduced for APOL1 kidney disease, this work should help in correctly assigning it to patients. Implementation should be easy and cost-effective since most commercial and noncommercial testing is based on gene panels, therefore the M1 variant is already captured and only needs to be reported.^41,42^ Moreover, in a kidney transplant setting, the knowledge of M1 would allow reclassification of APOL1-HR-M1 donors as nonrisk, thus expanding the donors pool. Similarly, this knowledge would improve risk assessment for kidney outcome of recipients of APOL1-HR-M1 kidneys. In asymptomatic individuals, detection of the M1 variant via genetic screening may indicate the absence of elevated susceptibility to APOL1-associated nephropathy and facilitate more precise risk stratification in population-level studies. A second goal of our study was to formally test the possible independent protective role of M1 in absence of APOL1-HR genotypes. Hence, we evaluated the difference in M1 prevalence among individuals with APOL1-LR genotypes across the same phenotypic categories and did not detect a protective effect of M1 for FSGS or SRNS nor for non-FSGS CKD as compared with control groups. This finding is consistent with the role of the M1 variant as a genetic modifier that exclusively neutralizes the deleterious effects of G2, without independently preventing non-APOL1 kidney disease. The fact that M1 did not impact individuals with a G0 allele is consistent with the well-established observation that G0 is the ancestral, nonrisk allele not associated with increased susceptibility to kidney disease. Furthermore, it suggests that APOL1-targeted therapies to prevent or treat kidney disease would not work for individuals who are only carrying LR G0- or G1-containing genotypes.
Limitations
This study has some limitations. First, the discovery cohorts include 8% individuals of African ancestry and 15% individuals of AMR ancestry. These numbers, while reflective of the prevalence of the genetic ancestries in the US population, might have negatively affected power, although our results reached statistical significance due to the enrichment of individuals with FSGS or SRNS and CKD in our discovery datasets. Second, our discovery cohort lacked granularity in age of onset and additional unmeasured clinical factors that could have influenced the presence of FSGS or CKD; nonetheless, our analysis focused on hard end points and relied on robust genotypic and phenotypic classifications that minimize the impact of missing clinical details. We also analyzed 3 large biobanks (eMERGE-III, AoU, and UKB) for validation. Our findings revealed a consistent direction of association with lower odds of having a diagnosis of FSGS or SRNS when carrying the M1 variant in individuals with APOL1-HR genotypes, though this difference did not reach statistical significance. These biobanks in fact include a much healthier population less enriched for kidney disease than our discovery dataset, especially for FSGS or SRNS. Nevertheless, it is notable that all individuals with APOL1-HR-M1 within the non-FSGS CKD group had ICD-9 codes that indicate clinical diagnoses unrelated to APOL1 kidney disease, such as diabetes, sarcoidosis, or systemic lupus erythematosus.
Conclusions
The findings of this case-control study suggest that APOL1 genotyping is incomplete without testing for M1. In fact, routine incorporation of M1 in the APOL1 genetic testing might enable physicians and genetic counselors to reclassify patients with an APOL1-HR genotype as not having kidney disease due to APOL1 and prompt the investigation for an alternative and potentially treatable cause of CKD not associated with APOL1. This will not only help improve diagnosis, prognosis, and treatment strategies, but may also avoid the possibility of substandard treatment for vulnerable populations, where ancestry and the presence of APOL1-HR genotypes might result in an unsubstantiated label diagnosis of APOL1 kidney disease with resulting incomplete diagnostic workup and inadequate care. Importantly, across biobanks in the US and Europe and across genetic ancestries, we confirmed the role of M1 as a genetic modifier only of the G2 allele, reinforcing the evidence that APOL1 G0 genotypes are not inherently deleterious. This study paves the way for increasingly personalized approaches to kidney disease diagnosis, management, and treatment, especially for individuals of African ancestry.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Udler MS, Nadkarni GN, Belbin G, . Effect of genetic African ancestry on e GFR and kidney disease. J Am Soc Nephrol. 2015;26(7):1682-1692. doi:10.1681/ASN.201405047425349204 PMC 4483587 · doi ↗ · pubmed ↗
- 2Kopp JB, Nelson GW, Sampath K, . APOL 1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22(11):2129-2137. doi:10.1681/ASN.201104038821997394 PMC 3231787 · doi ↗ · pubmed ↗
- 3Genovese G, Friedman DJ, Ross MD, . Association of trypanolytic APOL 1 variants with kidney disease in African Americans. Science. 2010;329(5993):841-845. doi:10.1126/science.119303220647424 PMC 2980843 · doi ↗ · pubmed ↗
- 4Tzur S, Rosset S, Shemer R, . Missense mutations in the APOL 1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH 9 gene. Hum Genet. 2010;128(3):345-350. doi:10.1007/s 00439-010-0861-020635188 PMC 2921485 · doi ↗ · pubmed ↗
- 5Friedman DJ, Pollak MR. APOL 1 nephropathy: from genetics to clinical applications. Clin J Am Soc Nephrol. 2021;16(2):294-303. doi:10.2215/CJN.1516121932616495 PMC 7863644 · doi ↗ · pubmed ↗
- 6Ojo AO, Adu D, Bramham K, ; Conference Participants. APOL 1 kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int. 2025;108(5):763-779. doi:10.1016/j.kint.2025.05.01740582702 · doi ↗ · pubmed ↗
- 7Egbuna O, Zimmerman B, Manos G, ; VX 19-147-101 Study Group. Inaxaplin for proteinuric kidney disease in persons with two APOL 1 variants. N Engl J Med. 2023;388(11):969-979. doi:10.1056/NEJ Moa 220239636920755 · doi ↗ · pubmed ↗
- 8Olabisi OA, Barrett NJ, Lucas A, . Design and rationale of the phase 2 baricitinib study in apolipoprotein L 1-mediated kidney disease (JUSTICE). Kidney Int Rep. 2024;9(9):2677-2684. doi:10.1016/j.ekir.2024.06.03339291185 PMC 11403079 · doi ↗ · pubmed ↗