Molecular detection of medically relevant Sporothrix species in roadkilled wildlife in the Brazilian Atlantic forest
Steffanie Skau Amadei, Julia Campos, Andressa Maria Rorato Nascimento de Matos, Keity Aparecida Speçato, Eloiza Teles Caldart, Ana Paula Frederico Rodrigues Loureiro Bracarense, Ferry Hagen, Zoilo Pires de Camargo, Anderson Messias Rodrigues

TL;DR
This study uses roadkill animals in Brazil to detect Sporothrix fungi, finding that wildlife may spread the zoonotic disease sporotrichosis.
Contribution
The study introduces roadkill as a novel biosurveillance tool for Sporothrix species and reveals new host associations and transmission dynamics.
Findings
Sporothrix DNA was detected in 13.6% of roadkilled wildlife samples.
S. brasiliensis was found in birds, challenging the thermal-exclusion hypothesis.
Reptiles were identified as potential hosts for Sporothrix species.
Abstract
The rapid expansion of zoonotic sporotrichosis in South America necessitates innovative surveillance strategies to identify natural ecological niches. Roadkill provides a unique, underutilized opportunity to monitor Sporothrix circulation within human-impacted landscapes. We conducted a molecular survey via a triplex probe quantitative real-time PCR (qPCR) assay targeting pathogenic Sporothrix species in 81 roadkilled vertebrates (mammals, birds, and reptiles) collected along highways BR-376 and PR-445 traversing the Atlantic Forest in Paraná, Brazil (2017–2023). Genomic DNA from visceral organs (heart, liver, lung, and spleen) was screened for fungal DNA. Sporothrix DNA was detected in 13.6% (11/81) of the samples. Sporothrix schenckii predominated, identified in wild mammals (Leopardus guttulus, Didelphis albiventris, and Lepus europaeus) and diverse birds (Colaptes melanochloros,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
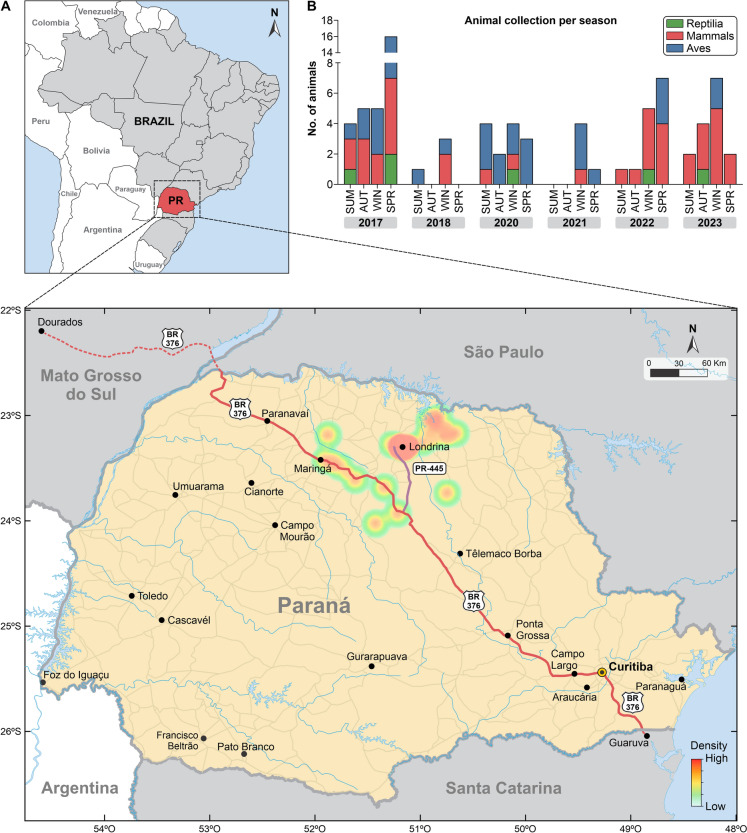 Figure 1
Figure 1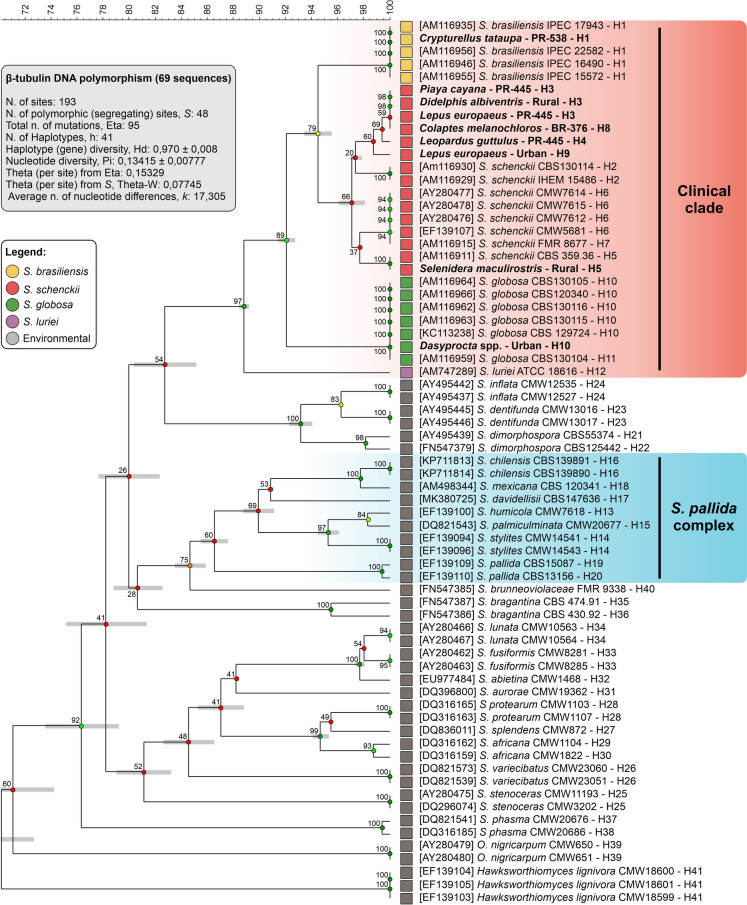 Figure 2
Figure 2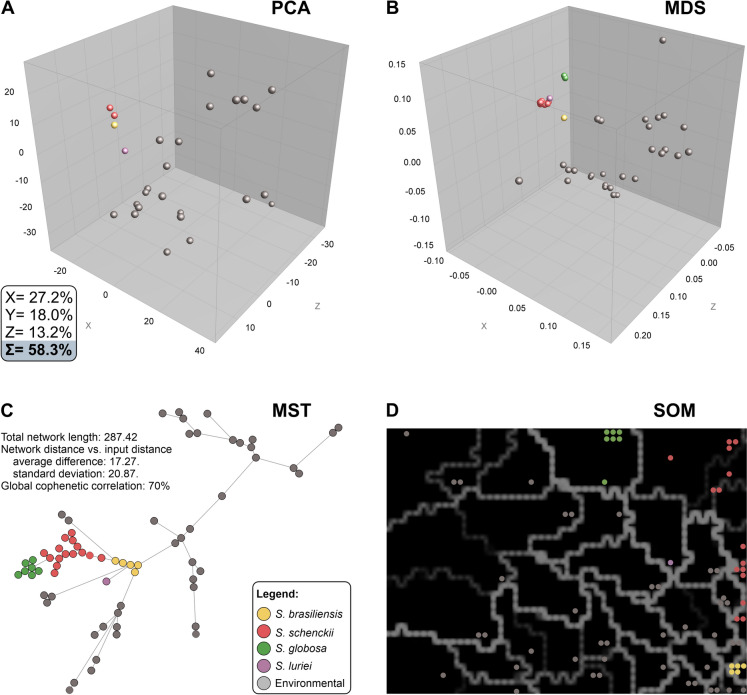 Figure 3
Figure 3- —https://doi.org/10.13039/501100001807Fundação de Amparo à Pesquisa do Estado de São Paulo
- —https://doi.org/10.13039/501100003593Conselho Nacional de Desenvolvimento Científico e Tecnológico
- —https://doi.org/10.13039/501100002322Coordenação de Aperfeiçoamento de Pessoal de Nível Superior
- —Universidade Federal De São Paulo
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFungal Infections and Studies · Parasitic Infections and Diagnostics · Leptospirosis research and findings
Introduction
Every second, approximately 15 wild animals are killed on Brazilian roads, totaling an estimated 1.3 million fatalities daily and over 475 million annually, which threatens biodiversity [1]. However, roadkill remains an underutilized resource for the surveillance of emerging zoonoses [2]. Roadkill samples provide valuable data for zoonotic surveillance, early detection, risk factor identification, mitigation strategy development, and investigations into pathogen ecological niches [3, 4].
Sporothrix (Ophiostomatales) encompasses thermally dimorphic fungi that cause sporotrichosis, a subcutaneous mycosis with increasing zoonotic recognition [5]. In Brazil, Sporothrix brasiliensis, S. schenckii, and S. globosa are of public health and veterinary significance [6]. Sporothrix brasiliensis is the primary agent of cat-transmitted sporotrichosis (CTS), driving outbreaks across Brazil and neighboring countries [7]; this contrasts with the global sapronotic transmission pattern associated with S. schenckii and S. globosa [6]. Brazil’s CTS-driven scenario focused epidemiological efforts on domestic cats (Felis catus), leaving the role of neotropical wildlife as reservoirs, mechanical vectors, or spillover hosts as critical gaps in the ecological understanding of Sporothrix [8].
To address these gaps, we employed real-time quantitative PCR (qPCR), a well-established, sensitive, and precise method for Sporothrix DNA detection [9]. The successful application of qPCR in roadkill studies for detecting bacteria [10], viruses [11], and parasitic agents [12] has reinforced its utility for pathogen surveillance. Sporothrix DNA was surveyed from roadkill collected along highways traversing the Atlantic Forest in Paraná, Brazil, a region with frequent interactions between wildlife, domestic animals, and humans. Our findings suggest that emerging Sporothrix species have active sylvatic circulation and broader host distributions, providing novel insight into the hidden role of roadkill in sporotrichosis ecoepidemiology.
Materials and Methods
Study Area and Biological Sampling
This study was conducted along the BR-376 (approx. 530 km through biodiverse forests/grasslands) and PR-445 (approx. 150 km through forest fragments/agricultural lands) highways in Paraná, Brazil, within the Atlantic Forest biome and the Campos Gerais region [13, 14] (Fig. 1).Fig. 1A Map of the study area in Paraná, Brazil, indicating the locations where 81 roadkilled animals were collected between 2017 and 2023. The sampling points are distributed along highways BR-376 and PR-445. B The collected fauna included mammals (n = 39), birds (n = 36), and reptiles (n = 6), and are detailed in Supplementary Table 1
The collection of roadkill involved weekly active surveys and notifications from law enforcement. Carcasses were retrieved within 24 h of collision and not eviscerated. Species identification and tissue collection followed the guidelines of Caldart et al*.* [15] under Brazilian wildlife research regulations (Ethics Committee on Animal Use, State University of Londrina, protocol no. 30/2017; SISBIO license no. 55384–1).
DNA Extraction and Quality Control
Genomic DNA was isolated from the collected tissue samples using the PureLink Genomic DNA Mini Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The concentration and purity of the extracted DNA were evaluated via a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). To confirm DNA integrity and exclude PCR inhibition, all samples were subjected to internal quality-control PCR targeting conserved vertebrate 12S and 16S rRNA sequences, as previously described [16]. Only samples in which vertebrate DNA was amplified were retained for pathogen screening.
Molecular Detection of Sporothrix spp. via qPCR
Samples were screened for medically relevant Sporothrix DNA via a triplex probe quantitative real-time PCR (qPCR) assay targeting β-tubulin polymorphisms to simultaneously detect and differentiate S. brasiliensis, S. schenckii, and S. globosa [9]. Reactions were performed in 20 µL containing 10 µL of TaqMan Genotyping Master Mix (Thermo Fisher Scientific), 900 nM of each primer Sporo-F and Sporo-R (Integrated DNA Technologies, USA), 250 nM of each species-specific TaqMan hydrolysis probe (Sbra-FAM, Ssch-NED, and Sglo-VIC; Thermo Fisher Scientific), and 5 µL of DNA template. Amplification was run on a StepOnePlus instrument (95 °C for 10 min; 45 cycles of 95 °C for 15 s and 60 °C for 1 min). The samples were considered positive when they presented an exponential amplification curve within the validated Cq detection range [9]. Each run included no-template and species-specific positive controls (Ss54 for S. brasiliensis, Ss126 for S. schenckii, and Ss06 for S. globosa) to confirm the reliability of the assay.
Amplicons generated from the Sporo-F and Sporo-R primers were purified via the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). To improve the quality of the sequencing data (Phred ≥ 30), the β-tubulin amplicons were subjected to control sequencing in two reactions. A BigDye Terminator v3.1 Cycle Sequencing Kit from Applied Biosystems was used for Sanger sequencing, and a SeqStudio Genetic Analyzer System was used to analyze the sequences (Applied Biosystems). Nucleotide sequences obtained from the β-tubulin amplicons were edited and assembled into consensus sequences using the CAP Contig Assembly Program implemented in BioEdit v. 7.0.9.0 [17].
Phylogenetic Analysis
To contextualize the roadkill-derived sequences, homologous reference sequences of Sporothrix spp. were retrieved from the NCBI GenBank database [18–21]. Phylogenetic analysis was performed using the BioNumerics software v.7.6 (Applied Maths, Sint-Martens-Latem, Belgium). Multiple sequence alignment was calculated using the fast algorithm with the following settings: an open gap cost of 100% and a unit gap cost of 0%. The fast algorithm parameters were defined with a minimum match sequence of 2 and a maximum number of gaps of 99. Similarity matrices were calculated on the basis of multiple alignment. The default cost table was used, with no correction applied for evolutionary distance. Cluster analysis was performed using the unweighted pair group method with arithmetic mean (UPGMA) method [22]. The robustness of the tree topology was evaluated via bootstrap analysis with 1,000 simulations [23]. In addition, the best-fit nucleotide substitution model was selected on the basis of the lowest Bayesian information criterion (BIC) score, implemented in MEGA11 [24]. The analysis indicated that the Kimura 2-parameter model with a discrete gamma distribution and evolutionary invariant sites (K2 + G + I) provided the optimal fit for the data. The Gamma-shaped parameter was estimated at 0.43, and the fraction of invariant sites was 25.0%. Phylogenetic relationships were also inferred using the maximum likelihood (ML) method, using a bootstrap analysis with 1,000 replications [23]. The initial tree for the heuristic search was obtained automatically by applying the neighbor-joining and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach. The final dataset included 69 nucleotide sequences with a total of 193 positions, following the complete deletion of all positions containing gaps and missing data.
Genetic Diversity and Haplotype Analysis
To elucidate the underlying genetic structure and reduce dataset dimensionality, principal component analysis (PCA) and multidimensional scaling (MDS) were employed. PCA and MDS utilized pairwise distance matrices in BioNumerics v.7.6 to identify the primary sources of variance, projecting the sequences into a three-dimensional coordinate space to visualize grouping behaviors and separation between taxa [25].
Complementary clustering and topology-based methods were applied in BioNumerics v.7.6 to further characterize sequence relationships. Minimum spanning trees (MSTs) were constructed to map the shortest evolutionary pathways between sequence types, illustrating the connectivity and distance between genotypes [26]. Additionally, self-organizing maps (SOMs) were generated via an unsupervised artificial neural network algorithm [27] to classify the entries on the basis of sequence similarity without a priori taxonomic assumptions. The SOM dimensions were optimized on the basis of sample size (determined by the heuristic formula \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$5.\sqrt{n}$$\end{document} ) [28], with the resulting spatial arrangement reflecting the degree of genetic relatedness among the studied populations [29].
Genetic Diversity and Haplotype Analysis
To assess the genetic variability of the identified Sporothrix populations, standard diversity indices were calculated via DnaSP software v.6.12 [30]. These metrics included the number of haplotypes (h), haplotype diversity (Hd), and nucleotide diversity (pi). To visualize the genealogical relationships and intraspecific divergence among the sequences, a haplotype network was constructed via the median-joining (MJ) algorithm implemented in Network software (Fluxus Engineering) [31].
Statistical Analysis
Epidemiological variables were tabulated and analyzed to identify factors associated with Sporothrix spp. detection. Categorical variables (taxonomic class, IUCN status, road type, season, proximity to urban centers, and interaction with domestic animals) were summarized as absolute and relative frequencies. Associations with qPCR positivity were tested via Pearson’s chi-square test (χ^2^) or Fisher’s exact test when the expected counts were < 5. All analyses were two-tailed, with statistical significance set at p < 0.05, and performed in GraphPad Prism v9.0.
Results
Between 2017 and 2023, 81 roadkill animals were collected: 39 mammals (48.15%), 36 birds (44.44%), and 6 reptiles (7.41%) (Fig. 1). A total of 178 tissue samples from the heart, liver, lungs, and spleen were collected (Supplementary Table 1). Medically relevant Sporothrix DNA was detected in 11 specimens (13.58%) across all three vertebrate classes (Table 1). Sanger sequencing provided orthogonal verification of the qPCR-positive samples (Supplementary Table 2). The tissues most frequently positive were the heart (n = 6) and liver (n = 5), suggesting potential primary infection or reservoir sites. Table 1. Detection of Sporothrix spp. DNA in road-killed wildlife from Paraná, Brazil (2017–2023)ClassSpeciesCommon nameCoordinates (Lat./Long.)RoadSeason/YearTissueqPCR (Cq)S. brasiliensisS. schenckii**S. globosaAvesColaptes melanochlorosGreen-barred woodpecker23°36′57″S51°18′36″WBR-376Spring/2017HeartN/D31,07N/DColumbina picuiPicui ground dove23°43′3″S51°23′47″WBR-376Spring/2017Liver42,78N/D18,71Crypturellus tataupaTataupa tinamou23°30′38″S51°14′3″WPR-538Winter/2017Heart41,54N/DN/DPiaya cayanaSquirrel cuckoo23°28′0.216′′S51°8′2.202″WPR-445Autumn/2020LiverN/D16,99N/DSelenidera maculirostrisSpot-billed Toucanet23°14′46″S51°56′22″WRuralSummer/2018LiverN/D26,07N/DMammaliaDasyprocta* spp.Agoutis23°20′47.0″S51°08′32.9″WUrbanAutumn/2023LiverN/DN/D42,09Didelphis albiventrisWhite-eared opossum23°21′16.6″S51°11′54.3″WRuralWinter/2023HeartN/D12,15N/DLeopardus guttulusSouthern tiger cat23°28′47′3″S51°7′57.8″WPR-445Summer/2020HeartN/D12,11N/DLepus europaeusEuropean hare23°18′49.9″S51°12′53.4″WPR-445Spring/2022HeartN/D35,38N/DLepus europaeusEuropean hare23°18′12″S51°10′15″WUrbanSummer/2017HeartN/D41,00N/DReptiliaOxyrhopus spp.False coral snakeN/AN/AUrbanWinter/2020LiverN/D18,2023,04^*^Cq, quantification cycle; N/D, not detected; N/A, not available
Statistical analysis revealed that qPCR positivity was associated with road type (χ^2^(3) = 9.640, p = 0.0219) and interaction with domestic animals (Fisher’s exact test, p = 0.0322) but not with other key variables, including taxonomic class (χ^2^(2) = 3.000, p = 0.2231), IUCN conservation status (χ^2^(1) = 1.000, p = 0.3173), season (χ^2^(3) = 4.000, p = 0.2615), or proximity to an urban center (χ^2^(1) = 1.000, p = 0.3173) (Supplementary Table 3).
The species most frequently detected was S. schenckii, which was found in eight samples from seven roadkilled animals, including the green-barred woodpecker (Colaptes melanochloros; heart, Cq 31.07), the squirrel cuckoo (Piaya cayana; liver, Cq 16.99), the spot-billed toucanet (Selenidera maculirostris; liver, Cq 26.07), two European hares (Lepus europaeus; hearts, Cqs 35.38 and 41.00), the southern tiger cat (Leopardus guttulus; heart, Cq 12.11), the white-eared opossum (Didelphis albiventris; heart, Cq 12.15), and the false coral snake (Oxyrhopus spp.; liver, Cq 18.20) (Table 1).
Sporothrix brasiliensis was detected in two bird species, the picui ground dove (Columbina picui; liver, Cq 42.78) and the tataupa tinamou (Crypturellus tataupa; heart Cq 41.54). Sporothrix globosa was identified in the agouti (Dasyprocta spp.; liver, Cq 42.09), the picui ground dove (Columbina picui; liver, Cq 18.71), and the false coral snake (Oxyrhopus spp.; liver, Cq 23.04). Notably, evidence of coinfection was observed in two cases in which the picui ground dove carried both S. brasiliensis and S. globosa DNA, whereas the false coral snake harbored S. schenckii and S. globosa DNA (Table 1).
The analysis of the BT2 gene fragment (69 sequences, 193 aligned sites) exhibited high genetic and haplotype variability, resolving 41 distinct haplotypes. Comprehensive polymorphism and nucleotide diversity metrics, indicating a substantial accumulation of genetic variation and divergent evolutionary history among the analyzed taxa, are summarized in Fig. 2 (inset) and Supplementary Table 4. The distribution of haplotypes was heterogeneous, with the majority being unique or shared by few individuals, suggesting active diversification. Notably, the roadkill-derived sequences were not randomly distributed but were consistently associated with specific, well-established genotypes described for the members of the clinical clade.Fig. 2UPGMA reconstruction of Sporothrix species recovered from roadkill wildlife on the basis of partial β-tubulin (BT2) gene sequences. The dendrogram was generated via BioNumerics v.7.6 (Applied Maths). Multiple sequence alignment was performed via the fast algorithm (open gap cost: 100%; unit gap cost: 0%; minimum match sequence: 2; maximum gaps: 99). Cluster analysis was performed via the unweighted pair group method with arithmetic mean (UPGMA), which is based on similarity matrices with no correction for evolutionary distance. The topology confirms that roadkill-derived sequences (highlighted) cluster strictly within the clinical clade (S. brasiliensis, S. schenckii, and S. globosa). Branch support values derived from 1,000 bootstrap simulations are indicated at the nodes. The GenBank accession codes and haplotype (H) numbers are close to each taxon. Although 11 samples were positive by multiplex qPCR, only nine sequences were represented in the phylogenetic tree because two cases involved coinfections (S. schenckii/S. globosa and S. brasiliensis/S. globosa; Table 1), which generated mixed Sanger chromatograms with ambiguous polymorphic sites, precluding reliable sequence assembly and phylogenetic inference (Supplementary Table 2)
Phylogenetic reconstruction and distance-based analyses confirmed that all amplicon sequences obtained from the roadkill samples fell strictly within the medically relevant clinical clade. The sequences clustered robustly with reference strains of S. brasiliensis (IPEC 16490; 100% ± 0.0%), S. schenckii (CBS 359.36; 97.1% ± 0.91%), and S. globosa (CBS 120340; 100% ± 0.0%) (Fig. 2), corroborating the species identification previously obtained via qPCR. This phylogenetic placement provides definitive evidence that the detected DNA belongs to pathogenic Sporothrix species and rules out the presence of environmental contaminants, nonpathogenic Sporothrix or Ophiostoma relatives in the positive samples (Fig. 2).
The median-joining haplotype network further elucidated the genealogical relationships, showing that wildlife sequences often shared haplotypes or were immediate neighbors to clinical and veterinary isolates described in previous studies. For example, S. brasiliensis sequences from the tataupa tinamou (Crypturellus tataupa) and picui ground dove (Columbina picui) clustered within the dominant S. brasiliensis haplogroup (H1), which is indistinguishable from known cat-transmitted strains (e.g., IPEC 16490 and IPEC 17943). Similarly, S. schenckii sequences from the squirrel cuckoo (Piaya cayana) and white-eared opossum (Didelphis albiventris) grouped in H3, demonstrating close genetic affinity with established lineages in H4 (CBS 359.36) and H5 (CMW 7614) (Supplementary Table 4).
To cross-validate the inferred population structure, multivariate statistical approaches, including PCA and MDS, in conjunction with topology-based clustering methods such as the MST and SOM, were applied (Fig. 3). In the PCA (Fig. 3A), the first principal component (PC1) clearly separates the medically relevant Sporothrix (S. brasiliensis, S. schenckii, and S. globosa) from the remaining taxa (environmental species), whereas the second principal component (PC2) further discriminates S. schenckii and S. globosa from S. brasiliensis. Together, the first three principal components (PC1–PC3) account for a substantial proportion of the total genetic variance, explaining 58.3% of the observed diversity. This ordination demonstrated that the roadkill-derived sequences clustered tightly with reference clinical isolates, whereas environmental species remained clearly segregated in the multivariate space (Fig. 3B).Fig. 3. Multivariate population structure and topological analysis of Sporothrix genotypes. A Principal component analysis (PCA) of genetic diversity; PC1 separates medically relevant Sporothrix species from environmental taxa, whereas PC2 discriminates between species within the clinical clade. The first three components explain 58.3% of the total variance. B Multidimensional scaling (MDS) plot corroborating the tight clustering of roadkill sequences with clinical reference strains. C Minimum spanning tree (MST) revealing the genealogical network topology; short branch lengths indicate high genetic affinity between wildlife isolates and established clinical genotypes (global cophenetic correlation = 70%). D Self-organizing map (SOM) analysis projecting roadkill sequences into the same topological neighborhoods as human and veterinary pathogens, which are strictly separated from environmental isolates
Consistent with the PCA and MDS results, the MST analysis revealed a compact and well-resolved network topology (Fig. 3C; model fit statistics detailed in the legend of Fig. 3). Short branch lengths and limited network expansion link wildlife-derived sequences directly to clinically relevant sequences, reinforcing the low genetic divergence among pathogenic genotypes. Reference sequences (environmental species) are shown in gray, providing a contextual framework for the positioning of the newly generated sequences. Finally, the SOM analysis (Fig. 3D) corroborated this distinct partitioning by projecting the roadkill-derived sequences into the same topological neighborhoods as the clinical reference strains, which were strictly separated from the environmental isolates.
Taken together, the congruence among phylogenetic inference (Fig. 2), haplotype networks (Supplementary Table 4), and multivariate clustering (Fig. 3) demonstrated that roadkill samples harbored sequences of S. brasiliensis, S. schenckii, and S. globosa. These findings strongly suggest the involvement of wildlife in the ecoepidemiology of sporotrichosis and highlight the robustness of the analytical framework, even when it is applied to short diagnostic genomic regions.
Discussion
This study provides novel molecular evidence for the presence of clinical Sporothrix spp. in roadkilled wildlife across ecologically diverse, anthropogenically influenced corridors in southeastern Brazil. Historically, the ecoepidemiology of sporotrichosis in Brazil was defined by its high prevalence in domestic animals, predominantly cats (Felis catus) [32] and dogs (Canis lupus familiaris) [33], with sylvatic reports largely confined to synanthropic rats (Rattus spp.) [34], coatis (Nasua nasua), capuchin monkeys (Cebus apella), and armadillos (Chlamyphoridae and Dasypodidae) [35]. We expand this established spectrum by detecting Sporothrix DNA in taxonomic groups previously unrecognized, such as reptiles, specifically the false coral snake (Oxyrhopus spp.); diverse native wild birds, including the picui ground dove (Columbina picui), tataupa tinamou (Crypturellus tataupa), green-barred woodpecker (Colaptes melanochloros), squirrel cuckoo (Piaya cayana), and spot-billed toucanet (Selenidera maculirostris); and mammals that act as potential bridges for visceral infection, including the southern tiger cat (Leopardus guttulus), European hare (Lepus europaeus), white-eared opossum (Didelphis albiventris), and agouti (Dasyprocta spp.).
These findings validate roadkill surveys as a unique window into hidden natural ecological niches, suggesting that sporotrichosis epidemiology extends beyond the classic sapronosis/zoonosis binary, mirroring patterns of other regional zoonoses [36–38]. Our data suggest that the Atlantic Forest acts as a pathogenic incubator under anthropogenic pressure, blurring the boundaries between sylvatic, rural, and urban transmission cycles [39]. The recovery of Sporothrix DNA from visceral organs across multiple vertebrates suggests that these animals may act as more than incidental hosts, potentially serving as reservoirs, biological amplifiers, or mechanical vectors [5].
Furthermore, the detection of Sporothrix DNA in peri-urban adapted species (Lepus europaeus, Leopardus guttulus, and Didelphis albiventris) points to their possible role as bridge hosts connecting sylvatic and synanthropic cycles [40]. Among these findings, the detection of S. schenckii in the heart of the vulnerable southern tiger cat (L. guttulus) is particularly noteworthy. Unlike cutaneous lesions typical of feline sporotrichosis [41], cardiac positivity implies hematogenous dissemination, indicating a threat of established systemic mycosis within wild felid populations.
Similarly, the detection of S. globosa in Dasyprocta spp. (agoutis) challenges the paradigm that this species is solely a low-virulence agent of fixed cutaneous infections [42, 43]. While armadillos (Dasypus novemcinctus and Tolypeutes matacus) are recognized as carriers owing to their digging behavior [44, 45], agoutis likely encounter inoculum through foraging and seed burial. Visceral detection suggests that under specific conditions, S. globosa can achieve dissemination [42]. Furthermore, the diversity of Sporothrix in wildlife, including members of the S. pallida complex found in other regions, such as S. davidellisii and S. humicola [18, 46], may represent a reservoir of genetic traits driving the evolution of pathogenicity within Ophiostomatales [47].
Our findings regarding S. brasiliensis in birds challenge the endothermy-exclusion hypothesis [48] and imply potential for long-distance aerial dispersal [49], complicating containment strategies. Ground-foraging birds, such as Columbina picui, may encounter Sporothrix propagules in contaminated detritus, particularly in ecotones, considering the high prevalence of this species in open and disturbed environments. Highly mobile birds (Columbina picui, Crypturellus tataupa, Colaptes melanochloros, Guira guira, Selenidera maculirostris, and Piaya cayana) may passively disseminate Sporothrix species propagules via avian excreta [49], a mechanism previously documented for mammalian feces [50, 51]. Furthermore, the documented transmission of S. brasiliensis from a cockatiel (Nymphicus hollandicus) suggests that birds can act as mechanical vectors [52].
The detection of Sporothrix in a false coral snake expands the known host spectrum of the pathogen [53]. This finding is consistent with outbreaks in free-ranging pygmy rattlesnakes (Sistrurus miliarius), from which *S. schenckii *sensu lato was isolated from skin lesions [53]. Reptiles, often neglected in mycological surveillance, may acquire infection trophically via rodent consumption [54] or through dermal contact, paralleling the ecology of Ophidiomyces ophiodiicola, the emerging agent of snake fungal disease [55]. Moreover, reptilian ectothermic thermoregulation might also select for fungal strains with rapid phase transitions, a key virulence factor in thermodimorphic Sporothrix species [56].
Current metagenomic evidence suggests that Sporothrix ecology is driven by biotic associations [57], whether with nematodes [58], insects [19], or mammals [8], rather than independent soil persistence [59]. This aligns with the “endozoan hypothesis” proposed for Coccidioides spp., which posits that the pathogen persists as an endozoan within the host and utilizes the carcass as a privileged, nutrient-rich substrate for saprobic proliferation and sporulation upon host death [60]. Although not statistically significant, the trend toward higher detection during the rainy season aligns with the Sporothrix thermohygrophilic profile [61], conditions that likely favor fungal proliferation within these Atlantic Forest microniches. With respect to tissue distribution, the detection of Sporothrix DNA predominantly in the heart and liver, rather than the lungs, argues against an inhalational route typical of other systemic mycoses, such as paracoccidioidomycosis [62]. Instead, this visceral pattern supports a mechanism of hematogenous dissemination following traumatic inoculation or trophic acquisition of infected prey or substrate.
Conclusion
We propose roadkill surveillance as a cost-effective and powerful tool for active monitoring of zoonotic fungal pathogens [10–12]. This strategy provides rapid access to elusive taxa (e.g., wild felids) and functions as an early warning system for environmental pathogen shifts, extending surveillance beyond the domestic cat reservoir [63]. In southeastern Brazil, a region heavily affected by the CTS, the detection of Sporothrix DNA across diverse taxa suggests that wildlife are not merely passive components of the ecosystem. While the molecular identification of S. brasiliensis in wild birds and S. schenckii in the hearts of wild predators highlights critical exposure and potential systemic dissemination, distinguishing between transient DNA carriage, true infection, and active disease will require future complementary histopathological studies.
Nonetheless, this multi-taxa molecular detection signals a significant risk of epidemic spillover into the broader ecosystem [5], underscoring the necessity of a One Health approach that transcends domestic animal control. Anthropogenic drivers, such as deforestation and urbanization, create ecological traps that foster pathogen exchange at forest-agriculture interfaces. Integrating molecular wildlife surveillance into national monitoring programs is therefore essential for predicting and mitigating future sporotrichosis outbreaks.
Supplementary Information
Below is the link to the electronic supplementary material.Supplementary file1 (DOCX 62 KB)