Early cellular plasticity promotes progression and dissemination in pancreatic adenocarcinoma
Giulio Innamorati, Giorgio Malpeli, Luca Giacomello, Roberto Salvia, Thomas M. Wilkie

TL;DR
This paper reviews how early cellular changes in pancreatic cancer drive its progression and resistance to treatment.
Contribution
The paper highlights the role of cellular plasticity in the early stages of pancreatic cancer development.
Findings
Phenotypic plasticity and EMT contribute to early cancer spread and treatment resistance.
Precancerous lesions like PanINs and IPMNs undergo molecular changes leading to malignancy.
Understanding these early events could improve therapeutic strategies and biomarker identification.
Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal malignancies, with limited therapeutic success and a persistently low 5-year survival rate. Despite significant advances in genomics and tumor biology, a fundamental challenge persists: to identify the elusive transformation from common benign pancreatic lesions to occasional malignant cellular identity. This review addresses a critical missing link in PDAC pathogenesis, focusing on when and where the switch to malignancy occurs, and why surgical intervention is often insufficient. We explore the biological and spatial–temporal evolution of precancerous lesions, such as PanINs and IPMNs, and examine how phenotypic plasticity and overlapping cellular programs—including squamous transdifferentiation, epithelial-to-mesenchymal transition (EMT), and acquisition of mesenchymal features—contribute to early dissemination,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
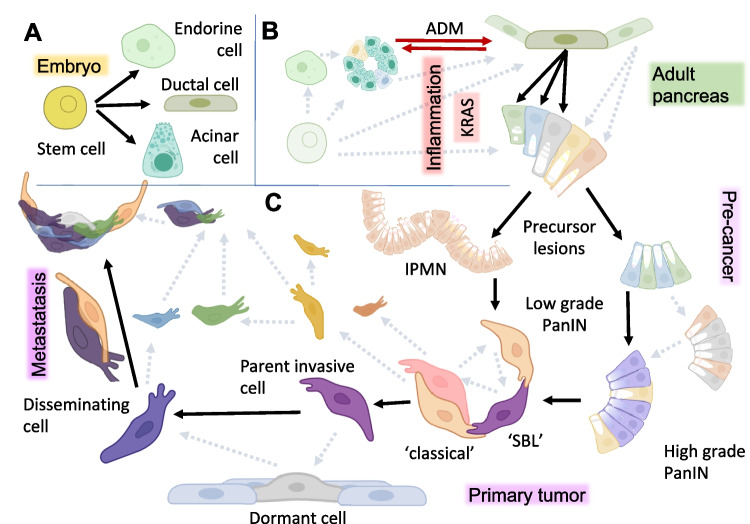 Figure 1
Figure 1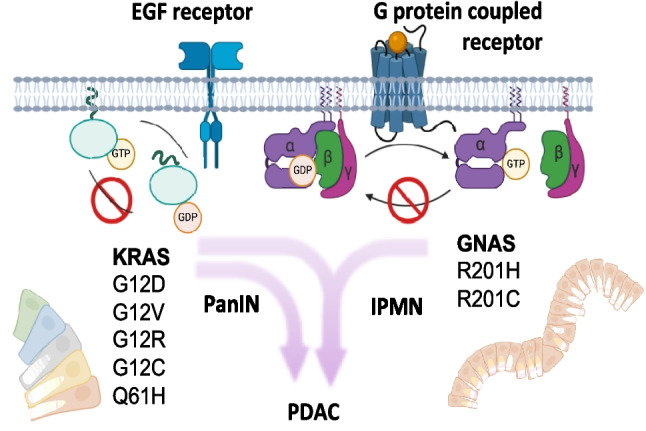 Figure 2
Figure 2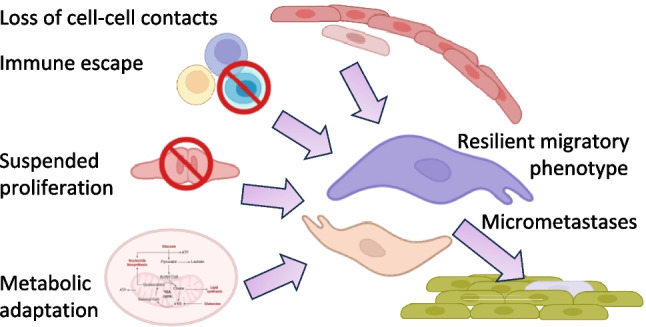 Figure 3
Figure 3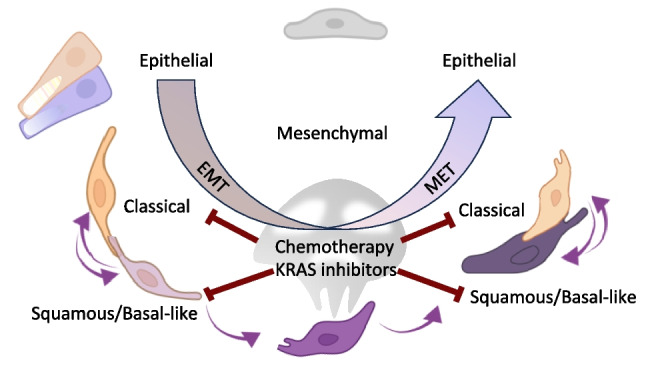 Figure 4
Figure 4- —https://doi.org/10.13039/501100005010Associazione Italiana per la Ricerca sul Cancro
- —https://doi.org/10.13039/100000054National Cancer Institute
- —Università degli Studi di Verona
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPancreatic and Hepatic Oncology Research · Cancer Cells and Metastasis · Single-cell and spatial transcriptomics
Introduction
In countertendency to most cancers, the global prevalence of pancreatic ductal adenocarcinoma (PDAC) is rising. This trend is expected to persist, driven in part by demographic changes such as longer life expectancy and the growing prevalence of obesity and diabetes mellitus [4]. Despite advances in cancer biology and therapeutics, progress in understanding PDAC initiation and improving patient outcomes remains limited. Early-stage PDAC is almost always clinically silent and most patients present with systemic disease involving lymph nodes, peritoneum, liver, or lungs. Even when tumors appear localized, pancreatectomy with negative margins rarely prevents recurrence [5].
These clinical patterns indicate that dissemination frequently occurs before diagnosis. Micrometastatic cells, invisible to current imaging modalities, evade both surgical removal and standard chemotherapeutic regimens. This raises fundamental questions about the earliest biological events that enable KRAS‑mutant epithelial cells—shaped by inflammatory cues, stromal interactions, and desmoplastic remodeling—to acquire plasticity enabling neoplastic transformation and early escape. Which molecular programs drive the progression of low‑grade precursor lesions into invasive disease, and which cellular populations within the tumor or its microenvironment are responsible for this silent spread? Defining the identity and timing of the cells that disseminate from very early pancreatic lesions—long before a detectable primary tumor forms—is critical for developing effective interventions.
Emerging spatial and high-throughput multiomic technologies have reframed PDAC as a dynamic, spatially organized ecosystem rather than a linear, cell-autonomous process. Clonal evolution with progressive accumulation of driver mutations alone cannot account for the transition from preinvasive lesions to widespread metastasis. Instead, integrated experimental approaches highlight the roles of epigenetic regulation, microenvironmental selective pressures, and epithelial–mesenchymal plasticity. Transient, reversible shifts between sedentary and motile stress-resistant states allow small subpopulations of cells to disseminate and persist as dormant or slowly cycling tumor cells. Spatial transcriptomic and proteomic analyses further reveal that invasive fronts and metastatic niches harbor distinct stromal and immune architectures, reflecting a branched evolutionary trajectory shaped by a continuum of adaptive cell states [6, 7].
In this review, we summarize recent advances and outstanding questions regarding the genetic and molecular mechanisms that drive early tumorigenesis, malignant transformation, and metastasis in pancreatic cancer, with particular emphasis on the role of phenotypic plasticity in tumor invasion and dissemination. Emerging data offer the potential to identify dispersed tumor cells that evade detection by conventional morphological or bulk analytical methods, ultimately opening the door to earlier prognosis and more effective therapeutic strategies.
Known drivers, obscure origins: the search for the metastatic progenitor in its provocative environment
Gain-of-function mutations in KRAS and subsequent loss-of-function mutations in tumor suppressor genes drive early tumorigenesis and subsequent progression in over 90% of PDAC [8]. However, recent advances in spatial biology and high-throughput omics technologies (reviewed in [3]) have challenged the traditional view that PDAC arises from a single clonal origin. Although pluripotent cancer stem cells are widely believed to drive disease dissemination, in PDAC their precise origins remain obscure, and no consensus has been reached regarding specific markers [9]. During embryonic development, three main lineages derive from pluripotent stem cells: acinar, ductal, and multiple endocrine subtypes (Fig. 1A). Pancreatic adult stem cells have long been suspected as progenitors for the three lineages as well as neoplastic cells; however, their identity, localization, and defining markers remain controversial [10]. An alternative hypothesis posits differentiated cells acquiring abnormal plasticity and reverting to a progenitor-like state [2, 11].Fig. 1PDAC origin—Black arrows represent accepted steps while grey dashed arrows describe less supported steps implying metachronous parallel evolution. Varying colors represent heterogeneity present at all levels. A During embryonic development, stem cells give rise to ducts, islets, and acini (green arrows). B In adult life, uncharacterized adult stem cells may restore differentiated cells. Acinar cells show heterogeneity and the highest degree of plasticity [2]. Inflammatory insults can induce a reversible transformation of acinar cells into ductal-like cells (ADM, red arrows). Persistent growth factor and CREB signaling or KRAS lock the duct-like cells in their transdifferentiated state promoting PanIN formation. Heterogeneity, exemplified by different colors, already present at the single-cell level in precancer lesions cannot be simply described by a phylogenetic tree ([3] and main text). Directly or indirectly, precancerous cells in PanIN or IPMN could originate from any pancreas cell lineages (dashed arrows). C The inactivation of other oncosuppressors further promotes progression. Among multiple independent precancerous lesions that accumulate in the aging pancreas, malignancy arises only rarely originating the primary tumor. When it does, early cancer development is characterized by heterogeneous cellular phenotypes and genotypes, without a clearly defined branching phylogenetic pattern. Sometime along this entire process, before clinical symptoms appear, unidentified cancer cells can disseminate beyond the pancreas, spreading as elusive and heterogeneous populations in lymph nodes, the liver, the lungs, the peritoneal cavity, and the bone marrow. Dormant cells within micrometastases may serve as a reservoir for cancer recurrence without ever becoming clinically apparent. The evolutionary process sees multiple subclones emerge early, compete for fitness, and differentially seed local invasion and distant metastases rather than following a single linear trajectory. Eventually, classical and SBL molecular phenotypes are preserved at the metastatic site
KRAS mutations (Fig. 2) and environmental insults can induce acinar-to-ductal metaplasia (ADM), a process in which acinar cells transdifferentiate into ductal-like cells. ADM is facilitated by inflammation and is reversible upon elimination of the inciting insult. The precise downstream mechanisms that drive the development of irreversible benign lesions remain undefined, including whether these lesions can arise from all pancreatic cell lineages (Fig. 1B) [2, 10, 12]. The prevalence of the most common pre-cancer formation, pancreatic intraepithelial neoplasm (PanIN), in the general adult population is estimated to be 60% to 86%, gradually accumulating in individuals over age 30 [13, 14]. Most precancerous lesions are driven by any one of several KRAS hotspot mutations that inhibit GTP hydrolysis, thus causing persistent activation. Analogous hotspot mutations in GNAS (Fig. 2) tend to develop intraductal papillary mucinous neoplasms (IPMN) which are larger, less aggressive cysts [15–17]. Three-dimensional spatial analysis revealed that grossly normal tissue can harbor hundreds of multifocal PanIN [18]. IPMNs are less common, with prevalence increasing with age and reaching about 10% in the general population over 50. A significant increase in PDAC development was observed only in patients with cysts classified as high risk according to Fukuoka criteria. Cysts without high-risk stigmata or worrisome features were associated with PDAC risk comparable to that of patients without cysts [19].Fig. 2. Oncogenic G-proteins driving PDAC. Highly specific hotspot mutations of G-proteins slow down GTPase activity, preserving and enhancing receptor downstream oncogenic signaling. Only a limited number of amino acid substitutions confer gain rather than loss of function. In pancreatic precancer lesions, GNAS mutations are specific for IPMN, often combined with KRAS, which is a driver in both PanIN and IPMN [1]
Both GNAS and KRAS driver mutations, due to the specificity of their base substitutions, are readily detectable by DNA sequencing in cancer tissue [20]. However, intratumoral heterogeneity in PDAC reflects diverse combinations of KRAS [21] and GNAS [22] mutations, implying that multiple precancerous lesions can arise independently and progress in parallel toward high-grade dysplasia. Morphological reconstruction has even revealed distinct KRAS variants within single PanINs, consistent with a multifocal intra-lesional origin rather than expansion from a single dominant clone [18]. One mechanism proposed to account for the metachronous emergence of such mutations is regional depletion of deoxynucleotide triphosphate (dNTPs) pools – for example, secondary to metabolic stress [23] – which promotes error-prone repair and genomic-wide mutagenesis, including oncogenes such as KRAS [24]. Crucially, these genetic events arise within a dynamic stromal and metabolic microenvironment that modulate cell state: nutrient limitation, extracellular matrix remodeling, and stromal signaling can foster phenotypic plasticity, enabling disseminating cells to adopt invasive or survival programs despite limited clonal expansion. Morphologic analysis suggests early lesions arise as interspersed neoplastic cells within apparently normal ductal cells and surrounding fibroblasts. As these lesions grow and branch, they displace acinar cells, entrap islets, and encircle peripheral nerves. Concurrent pancreatitis contributes an inflammatory cicatricial matrix, typical of the tumor microenvironment and variably populated by fibroblasts, lymphocytic, and myeloid infiltrate [25]. Cancer cells are marked by oncogenic mutations but not the stromal cells of the tumor microenvironment. If GNAS and KRAS mutations represent early events, then genetic heterogeneity emerges during the initial phases of transformation, and PDAC appears to evolve over a diffused network of multiclonal and convoluted PanINs. Together, these observations link multifocal mutagenesis and microenvironmental pressures to the emergence of phenotypically plastic cells capable of early dissemination. The overall scenario sees the tumor mass generated by a mosaic of precancer cells with distinct signaling properties. Subsequent mutations in tumor suppressors (e.g. TP53, SMAD4, CDKN2A) and copy number variation (CNV) alterations drive toward malignant progression.
Marked for departure: spotting the pre-metastatic pioneers
Studies using single-nucleotide variants and insertions/deletions to analyze microdissected PanINs and distant PDAC lesions within the same pancreas [26] have revealed:
- Distinct progeny suggesting synchronous growth.
- Close proximity when PanINs share all driver mutations.
- Common ancestors indicating branching during progression.
These findings suggest that multiple precancerous cells, morphologically appearing as PanIN and already carrying the most common driver mutations, spread and progress through ducts [26]. This raises concerns about the specificity of early diagnostic tests based solely on KRAS or GNAS mutations, as these alterations are also found in benign precursor lesions. In fact, mutant DNA has been detected in the plasma of healthy individuals and patients with pancreatitis [27]. In addition to these potential confounding effects, a blood-based diagnostic biomarker that detects PDAC at a surgically curable stage must identify cancer when lesions are still subcentimeter. At this stage, the mass will contain relatively few tumor cells, show minimal necrosis, and thus release a very small amount of cell free DNA only modestly higher than in advanced precursor lesions. Even with current high-performance circulating tumor DNA assays, at such an early stage, the probability of detecting tumor-derived circulating DNA in peripheral blood is very low because mutant fragments are highly diluted within the total cell-free DNA pool. Detection of actively secreted proteins or small vesicles are expected to be more sensitive reporters. At present, biomarkers such as Ca19.9 are only informative for monitoring disease progression once a significant tumor burden is established within the pancreas and metastatic sites [28]. Although it is widely accepted that cancer cells can enter lymphatics vessels or the bloodstream, the precise identity and mode of transit of these cells remain poorly defined: it is unclear whether they disseminate as single cells or as multicellular clusters; it has even been proposed that dissemination may involve fusion between tumor cells and macrophages [29]. Consequently, research should prioritize detailed characterization of early intraductal dissemination and the development of highly sensitive biomarkers capable of detecting subcentimeter, surgically curable lesions.
The struggle of tracing ancestors of disseminated cells
Characterizing asymptomatic progression to systemic disease represents a major challenge for researchers since authentic early-stage human samples—particularly from the pancreas—are virtually inaccessible. The collection of human tissue samples either comes from advanced cases or from autoptic material that can only provide incidental pre-neoplastic lesions that only in rare and unpredictable cases may have evolved to cancer during subsequent years. As a result, most research has relied on tissue specimens derived from samples of clinically manifest illness collected from patients who underwent surgical procedures, leaving the timing and cellular origin of malignancy substantially unresolved. Resected specimens inevitably fail to accurately represent the early-stage disease from which disseminating cells originated.
Two methodological approaches may overcome this issue. One is a periodic collection and storage of samples and clinical information from a large population of ‘healthy individuals’. Rare cases developing the disease will have available a historical set of clinical data and samples. A similar massive effort to cover the history of PDAC insurgency from the origin is being undertaken by the PRECEDE consortium (www.precedestudy.org) focused on high-risk individuals limited to relatively small plasma aliquots, or other specimens that can be collected by non-invasive procedures, lacking early tissue samples.
Otherwise, much like archaeologists, researchers must reconstruct the phylogenesis of the disease, delineating—at the single-cell level—the processes of de-differentiation and re-differentiation that drive malignant progression. CNV analyses of matched primary tumors and metastases across multiple organs in the same patient have failed to reveal a unified evolutionary trajectory. Instead, multiple phylogenetic structures have been inferred, indicating complex and branched patterns (Fig. 1C) of cancer cell dissemination [30].
Recent advances, particularly in single-cell analysis and spatial biology, have revealed that PDAC is a highly heterogeneous and multifocal disease within individual patients [31–33]. Perhaps for this reason the efforts to personalize therapy based on mutational profiles have not yielded the anticipated clinical benefit [34]. The fateful step and timing of when disease disseminates remain to be clarified.
Transformed cells leave home: the escape from primary tumors
If in the context of the acquisition of cellular plasticity, as a seminal event for cancerogenesis, genomic analysis has failed to yield a comprehensive model of PDAC onset and progression. Recent advances have emerged from efforts to characterize the intricate crosstalk between gene transcription and the extracellular microenvironment. Among these, EMT and inflammation are recognized as early and interrelated events in tumorigenesis [35]. Microenvironment-induced EMT promotes epithelial cell migration and contributes to cellular transformation and invasiveness, thereby playing a pivotal role in the initial steps of dissemination. In the earliest stages of pancreatic neoplasia, PanIN lesions arise within a stroma enriched in nerve fibers and collagen deposition, which progressively evolves into the dense desmoplastic matrix that defines PDAC histopathology (reviewed in [36]). Importantly, the structural properties of collagen I differ depending on its source: fibroblasts mainly synthesize the heterotrimeric isoform, whereas transformed epithelial cells produce the homotrimeric variant. These differences critically modulate immune cell recruitment and enhance invasive behavior [37].
Similar to human disease, in most genetically engineered mouse models (GEMMs), PDAC initiation depends on orthotopic expression of KRAS^G12D^. Acinar cells expressing KRas isolated from murine pancreas and embedded in collagen initiate ADM under low-stiffness conditions. Yet, they reprogram transcription to express ductal/progenitor markers only upon increasing rigidity after YAP/TAZ nuclear translocation [38]. Tumor progression accelerates when KRAS^G12D^ expression is combined with loss of tumor suppressors such as Tp53 (KPC model) and/or Ink4a (KIC model). Although these models artificially shorten early disease stages and may not fully capture the genetic heterogeneity of human PDAC—particularly that of disseminated tumor cells [39]—they demonstrate that inhibiting collagen-fibers deposition can prevent neoplastic lesion formation [38].
Mechanistic studies in GEMMs further indicated that EMT and cell dissemination occur surprisingly early, before the appearance of overt carcinoma. KRAS-positive PanIN or ADM cells have been observed to delaminate and enter the bloodstream, consistent with early systemic spread [40]. More than a decade ago, lineage-tracing experiments in KPC mice revealed that pancreatic-derived mesenchymal-like cells colonize distant organs such as the liver long before histological detection of malignancy. Blocking inflammation in these models prevented PanIN formation, suppressed EMT, and abolished early cell dissemination [35]. A more recent study introduced a TFF1 knockout in the KPC background to enhance EMT features [41]. Tagged cells disseminated more efficiently at advanced PanIN or early PDAC state than later stages, supporting the hypothesis formulated for other malignancies, particularly for ductal carcinomas (breast, melanoma, esophageal, colorectal, ovarian), that neoplastic cells can spread early and remain dormant and undetected at distant sites for several years before clinical manifestation [42]. Remarkably, disseminated cells exhibited phenotypic plasticity and even expressed tissue-specific markers, resembling hepatocytes in the liver, pulmonary epithelium in the lung, and leukocytes in the bone marrow [41]. Complementary GEMM, designed to distinguish EMT-proficient epithelial cells from those acquiring a mesenchymal phenotype, confirmed that mesenchymal plasticity occurs in PanIN. In this model, the activation of EMT program results in genomic instability, chromothripsis, and the acquisition of metastatic competency [43]. Thus, PDAC dissemination is likely to occur long before the formation of a solid mass.
Clinically, parallel evidence supports early systemic spread in patients. While bone metastases are uncommon, extensive studies demonstrated early dissemination of neoplastic cells in the bone marrow, often carrying a reduced burden of driver mutations [42]. KRT7/8-positive cells have been detected in roughly one-third of cases and correlate with poorer prognosis and smaller yet more aggressive primary tumors [44]. These findings suggest that dormant micrometastases, seeded during the earliest phases of tumorigenesis, may persist in quiescent niches. Though clinically silent, such lesions can shed cells that ultimately undermine the efficacy of surgical resection of tumors deemed localized [45].
The migratory phenotype: hallmarks of cellular journey
Depending on microenvironmental cue and cell-state transition, PDAC cells can employ different migration strategies including collective, mesenchymal, and amoeboid migration [46]. EMT endows cells with the ability to migrate, survive under stress, and evade immune surveillance, and may not form a manifest tumor mass. EMT adaptation involves several key and interdependent changes for dissemination (Fig. 3):
- Loss of cell–cell adhesion and reorganization of junctional complexes, cytoskeletal remodeling, and microenvironmental interactions are required to migrate from the primary tumor site. Moving through dense ECM is highly energy demanding, with rapid ATP turnover at actomyosin structures and focal adhesions. In addition, cells can either migrate individually or collectively following leader cells. Higher energy costs paid by the leader cells are compensated by transient upregulation of glycolysis or even genetic modifications to upregulate oxidative phosphorylation [47].
- Metabolic reprogramming to support movement and survival under adequate oxygen conditions or nutrient- and oxygen-deprived environments. Reframing the concept of the Warburg effect, PDAC cells can exploit glycolysis-high and OXPHOS-high states to reconfigure glucose, glutamine, lipid, and redox pathways in response to mechanical, nutrient stresses or drug exposure [48]. As a result, lactate resulting from glycolysis can trigger signaling further supporting survival, invasion, metastasis, angiogenesis and immune escape [49]
- Alteration of surface epitopes and immune escape by multiple mechanisms reviewed in [50] includes acidosis derived from glycolysis which inhibits effector cells such as CD8 + T and NK cells, while supporting immunosuppressive regulatory T cells and myeloid-derived suppressor cells [51]. Fig. 3EMT and dissemination. Epithelial cells undergo loss of tight junctions and adopt migratory stress-resistant phenotypes that permit dissemination and subsequent engraftment at distant sites, where new contacts are formed. In PDAC, occult micrometastases maintain disease dormancy. Later reactivation produces liver and peritoneal metastases, which are the principal drivers of morbidity and mortality
- Modulation of proliferation, with reduced division during migration (Fig. 3).
In PDAC, these features are characteristic of the squamous/basal-like (SBL), the molecular subtype identified in the attempt to stratify patients at the transcriptomic level [52].
Diverging destinies: the “classical” and “squamous” progeny
Adenocarcinoma or squamous cell carcinoma are the two major histological subtypes of epithelial carcinoma. However, adenocarcinoma can acquire squamous properties. In PDAC, pathologists have long noted a squamous cell component in a subset of tumors. When this component exceeds one-third of the tumor mass, the diagnosis is revised to adenosquamous carcinoma, a variant associated with increased aggressiveness. Proteomic comparison across distinct tumor types has identified transversal markers that distinguish squamous carcinoma from adenocarcinomas cells irrespective of tissue origin [53]. This suggests a shared transcriptional reprogramming that drives the transition from an epithelial to a squamous phenotype.
Unsupervised transcriptomic analyses aimed at finding prognostic molecular signatures produced multiple subclassifications of gastrointestinal malignancies. Across these efforts, a mesenchymal molecular pattern consistently overlapped with poorer prognosis relative to epithelial subtypes [54].
In PDAC, molecular taxonomy evolved from early three‑subtype schemes in the 2010 s to more refined multi‑subtype classification with advances in genomics, transcriptomics, and single‑cell technologies. These definitions have eventually converged into two major transcriptomic subtypes: the classical and the SBL (reviewed in [55] which also explains how additional subtypes may represent contaminations from other lineages or intermediate stages). KPC mouse models indicate that mutations in acinar cells preferentially give rise to the classical subtype, whereas SBL subtype cells would arise from ductal cells [56]. However, single-cell analyses have shown that both subtypes coexist within the same tumor, contributing to intratumoral heterogeneity, with the SBL component associated with poorer prognosis and greater metastatic potential [3, 57].
Microdissection of morphologically distinct lesions (200–500 cells) by Di Chiaro et al. identified three "morpho-biotypes": gland-forming, non-gland-forming, and poorly differentiated variants [28]. RNA analysis linked these morphologies to molecular subtypes: gland-forming lesions were predominantly classical-like, while poorly differentiated and signet-ring cell–containing lesions were enriched for SBL features [58].
The SBL subtype exhibits several traits associated with aggressive disease, including:
- Immune evasion;
- Adaptation to hypoxia, even under normoxic conditions;
- Enhanced metastatic potential;
- Therapy resistance;
- Worse clinical outcome [59].
Although EMT has been reported to be activated in both classical and SBL subtypes, the underlying transcriptional programs differ [59]. The squamous transcriptional pattern is characterized by abundant extracellular matrix synthesis and EMT, consistent with a migration-prone phenotype. The GEMM study referenced above, designed to dissect EMT dynamics, indicates that EMT facilitates the acquisition of mesenchymal traits along with progressive genomic instability, resulting in genomic aberrations and elevated transcriptional entropy; conversely, EMT inhibition reduces phenotypic heterogeneity and preserves ductal-like morphology and classical-like transcriptional profile. Thus, EMT contributes to generating multiple transcriptomic trajectories that enhance tumor heterogeneity [43]. It is therefore plausible that the SBL phenotype reflects an EMT-driven evolution of a high-fitness population with increased metastatic potential and therapy resilience.
Despite this, transcriptional profiling by Park et al. [59] and Pei et al. [30] reveals both classical and SBL components persist in metastatic lesions, with the classical subtype cells showing only a relative preference for lung colonization over liver metastasis [30]. These findings suggest that expression subtype acquisition is not tied to the clonal architecture inferred from genomic analysis and that the relative capacity of each subtype to disseminate remains incompletely understood. Aiello et al. associated the classical phenotype with a partial EMT program and SBL with an alternative, more complete EMT program that—through transcriptional regulation—drives single-cell migration of SBL cells [60]. The same lineage-labeled KPC model indicates that the classical subtype loses the epithelial characteristics by internalizing membrane proteins such as E-cadherins. By retaining intercellular cohesion, tumor spheres derived from classical cells were reported to invade a 3D matrix via collective cell migration, a process recently shown to be facilitated by aligned collagen, which reduces the energetic demands for migration in PDAC BxPC-3 cell line [61].
An additional layer of complexity arises from the need for disseminated cells to adapt to the local microenvironment, which can obscure their origin. Spatial adaptation of PDAC cells has been reviewed in detail by Maddipati, including implications for therapy [62]. Even at sites where metastases are not commonly detected, engrafted cells can acquire organ‑specific genes, as mentioned above for most common metastatic sites (liver and lungs) [63]. For example, PDAC cells infiltrating the duodenal wall have been shown to respond to local cues, integrating into the native epithelium and adopting classical phenotypic features while preserving the mucosal architecture [64]. Reproducing the true complexity of pancreatic cancer cell spread in experimental models is evidently a formidable challenge.
Cultivating complexity—why organoids fall short as patient stand-ins
While the transition between classical and squamous lineages defines a clinical trajectory of the disease, our ability to model this plasticity remains limited; even our most advanced 3D organoids often fail to capture the TME-driven signals that trigger these lineage shifts in the patient. The relevance of PDAC experimental models (including combinations of 3D, slice cultures, tumor-on-a-chip, and animal models) to orient precision medicine strategies was recently reviewed in ref. [65]. Patient-derived xenografts obtained from tumor fragments (PDX) or tumoroids (PDO) are widely used and currently the best models for personalized therapies. However, inevitably, murine avatars are established using cells harvested from the primary tumor post-diagnosis, at a point when the malignancy has already metastasized beyond the tissue of origin. Consequently, their fidelity in recapitulating the tumorigenic inception or in forecasting systemic eradication of neoplastic cells remains unvalidated. Furthermore, xenografted human cells disseminate within the murine host in the absence of critical microenvironmental elements, including the dysplastic extracellular matrix, stromal fibroblast populations, and the patient's autologous immune response. The inevitable loss of the original microenvironment in ex vivo models dramatically affects heterogeneity.
From a clinical implementation standpoint, turnaround time remains a major bottleneck: the establishment, expansion, and pharmacotyping of patient‑derived PDAC organoids often exceed the clinical window available for first‑line treatment decisions. Nevertheless, the field is rapidly advancing its ability to preserve tumor complexity [66] and is expected to ultimately inform and improve therapeutic strategies.
Single-cell analysis of tumors derived from a modified version of the KPC mouse model suggested a continuum going from the classical transcriptional subtype, enriched in mucins, towards mesenchymal transitioning through a basal-like intermediate state enriched in keratins and laminin. Two-dimensional cell culturing skews towards the BSL phenotype as opposed to 3D organoids culture. However, plasticity was conserved, as in both models heterogeneity could be restored by orthotopically implanting cells in NSG mice [67]. Organoids studies have provided further insights into subtype plasticity. Early-stage organoids display cystic morphology, Wnt-dependence, and classical transcriptomic features whereas advanced organoids exhibit Wnt-independence and express squamous markers such as TP63, KRT5, and KRT6A [68]. Standard organoid culture conditions include Wnt3a which promotes the canonical signaling pathway modulating βcatenin regulated transcription. In contrast, non-canonical Wnt ligands, such as Wnt7a, promote the non-canonical pathway mediated by planar cell polarity and Ca^2+^ signaling upstream transcriptional programs associated with motility and metastasis. Autocrine production and the inner core of a filled organoid could recreate the conditions for SBL cells. This suggests that current protocols based on Wnt3a, R-spondin, epithelial differentiating agents like noggin, and normoxia may facilitate the classical phenotypes, favoring initial proliferation, gland-forming morphology, and underrepresenting the SBL subtype [69, 70]. Conversely, within the hypoxic inner core of a filled organoid, it is plausible to hypothesize that autocrine factor production, coupled with minimal exposure to exogenous bovine serum components, more accurately recapitulates the microenvironmental conditions conducive to the emergence of the SBL phenotype [71] and cell heterogeneity. Nonetheless, such culture conditions are unlikely to reproduce the full spectrum of molecular divergence in systemic disease from which the patient-derived organoid originated [72]. Improving culture models to manipulate transdifferentiation of the classical and SBL subtypes remains challenging, particularly due to the difficulty in replicating microenvironmental cues, including mechanical tension that has been implicated in triggering malignant transformation [73].
Targeting the shifting target: how plasticity and subtyping could redefine therapy
While the precise relationship between classical and SBL subtypes remains unresolved, several scenarios are plausible. The SBL phenotype may arise through subclonal evolution from classical, glandular tumors [74]. Alternatively, the two subtypes may originate independently yet co-evolve and co-disseminate, potentially under the influence of SBL lineages; for instance, ΔNp63α—a marker and driver of squamous differentiation and the SBL phenotype—has been shown to promote EMT and collective migration in basal-like breast cancer [75]. Finally, the balance between classical and SBL dissemination may simply reflect plasticity driven by multiple factors, including passaging, oxygen tension and loss of cancer-stromal cell interactions within a microenvironment that changes during dissemination, dormancy outbreak or co-evolve in the primary site [30, 76] First-line systemic regimens are FOLFIRINOX (folinic acid, 5-fluorouracil, irinotecan, oxaliplatin) and gemcitabine plus nab-paclitaxel with a similar overall survival benefit between the two regimens. According to the COMPASS study, locally advanced or metastatic classical tumors responded better to modified FOLFIRINOX than basal-like, which may have a relatively better response to gemcitabine/nab-paclitaxel. However, only 20% of patients were “basal-like” and all at stage IV; the impact of differentiating treatment based on classical vs. SBL subtypes remains extremely limited [55, 77]. On the other hand, molecular profiling may become essential in positioning treatment with novel drugs that target KRAS and its downstream signaling. Great expectations are raising with the development of MRTX1133, which targets the G12D mutation predominant in PDAC, whereas G12C predominates in lung cancer and warrants the use of Sotorasib and Adagrasib. Unfortunately, a major limit to KRAS inhibition efficacy is the rapid emergence of drug resistance [78]. Secondary mutations in KRAS or other drivers are found involved in a limited number of cases. Given the relatively short temporal window of resistance development, these mutations likely exist prior to first round treatment, and cellular lineage plasticity adds to subclonal heterogeneity associated with compensatory reprogramming of signaling and metabolic pathways [79, 80]. Recent data from mouse and human PDAC samples revealed BSL cells are more sensitive to KRAS inhibition, compared to classical cancer cells, which appeared to become the reservoir for disease relapse [80, 81] (Fig. 4).Fig. 4. Vulnerability of a dynamic phenotype. Classical vs. BLS largely map onto epithelial vs. mesenchymal ends of the EMT–MET spectrum, but both signatures sit on a shared, plastic continuum enriched for hybrid (partial EMT) states rather than two discrete and non-overlapping programs. In practice, BLS signatures are strongly EMT-skewed, while classical signatures are epithelial/MET-skewed, with single cells co-expressing elements of both programs. An appropriate combination of therapies could reveal more effective targeting SBL and classical
If BSL and classical are two faces of the same medal, treatment options should consider both within a timeframe that reduces the expansion of drug-tolerant cell populations. In this perspective, a practical classification of tumor subtypes in clinical settings becomes key. Feasibility barriers in PDAC molecular subtyping arise from the high cost, limited accessibility of comprehensive real-time genomic profiling, and viable biopsy availability. Immunohistochemistry is limited by inherent subjective variability, poor cancer cellularity, and only EUS will be available for neoadjuvant therapy. To circumvent these issues and develop a standardizable approach for implementing PDAC subtype classification, the Purity Independent Subtyping of Tumors (PurIST) algorithm was obtained by machine-learning–based, single-sample classifier. It evaluates relative expression ratios across eight gene pairs derived from tumor-intrinsic genes [46]. A test based on pancreatic tissue appears as feasible and clinically meaningful for advanced, treatment-naïve patients. Monitoring cell plasticity and the efficacy of treatment schemes designed to tackle the disease on multiple fronts would strongly benefit from noninvasive and repeatable tests.
Conclusion
Extensive efforts to refine molecular signatures specific to malignant cells have led to the adaptation of concepts such as EMT, mesenchymal identity, and squamous differentiation in PDAC. These overlapping features reflect the remarkable cellular plasticity of PDAC, underlie the poor therapeutic response, and highlight the need for a clear understanding of tumor evolution. Recent advances in spatial biology and single-cell analysis suggest a paradigm shift is needed—one that prioritizes identification and treatment of asymptomatic disease and micrometastases.
Historically, research has focused on tissue samples and PDAC models that refer to advanced stages of disease within the pancreas. It remains difficult to obtain human tissue samples representative of the asymptomatic phase, between the preinvasive lesions and the authentically localized tumor. While GEMMs remain one of the most informative tools for studying early PDAC progression, their reliance on specific driver mutations fails to fully capture the genetic and phenotypic complexity of human disease. The limitations of GEMMs have been highlighted by the repeated failure of clinical trials based on preclinical findings from these models, particularly in recapitulating the full spectrum of metastatic behaviour [82].
The rapid evolution of three-dimensional models, such as organoids and tumoroids, offers the potential to incorporate patient-derived precancerous and stromal cells to better reflect the complexity of immune interactions and stromal dynamics. When integrated with spatial biology insights and advanced in vivo models, like GEMMs and PDXs, these systems provide a powerful framework for decoding cellular phenotypic fluidity.
This multimodal approach is essential for understanding asymptomatic dissemination and eradicating the micrometastases that currently limit the efficacy of surgical interventions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jimenez-Sanchez, A., Persad, S., Hayashi, A., Umeda, S., Sharma, R., Xie, Y., Mehta, A., Park, W., Masilionis, I., Chu, T. et al. (2025). Transcriptomic plasticity is a hallmark of metastatic pancreatic cancer. Cancer Research. 10.1158/0008-5472.CAN-25-111710.1158/0008-5472.CAN-25-1117 PMC 1304453241379552 · doi ↗ · pubmed ↗