Case Report: KMT2A amplification in two adult patients with B-cell acute lymphoblastic leukemia
Min Gao, Yunjia Chen, Kimo Bachiashvili, Pankit J. Vachhani, Omer Jamy, Shuko Harada, Alexander Craig Mackinnon, Nirupama Singh, Aishwarya Ravindran, Baleed Vishnu Reddy, Andrew J. Carroll, Fady M. Mikhail

TL;DR
This case report describes two rare adult cases of B-cell acute lymphoblastic leukemia with KMT2A gene amplification, highlighting their poor prognosis and genetic features.
Contribution
The paper presents two new adult cases of KMT2A-amplified B-ALL and reviews existing literature to emphasize its clinical significance.
Findings
KMT2A amplification in B-ALL is rare and associated with poor outcomes in older adults.
TP53 mutations and CRLF2 rearrangements are frequently observed in KMT2A-amplified B-ALL cases.
Genetic profiling is critical for risk stratification and treatment of KMT2A-amplified B-ALL.
Abstract
The KMT2A gene, located at chromosome band 11q23, encodes a lysine methyltransferase essential for hematopoietic gene regulation. While KMT2A rearrangements are common in acute myeloid leukemia (AML) and B-cell acute lymphoblastic leukemia (B-ALL), KMT2A amplification is rare, occurring in ~1% of AML cases and even less frequently in B-ALL. Given its rarity, understanding KMT2A amplification in B-ALL is crucial for improving diagnostics and therapy. We report two adult B-ALL cases with KMT2A amplification. Patient 1, a 58-year-old male, had KMT2A amplification (6~18 copies in 68.5% of bone marrow cells), a complex karyotype, and a pathogenic TP53 variant (c.524G>A, p.Arg175His). He underwent induction chemotherapy but passed away after two months due to complications. Patient 2, a 66-year-old female, had KMT2A amplification (8~11 copies in 87.5% of peripheral blood cells) and CRLF2…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
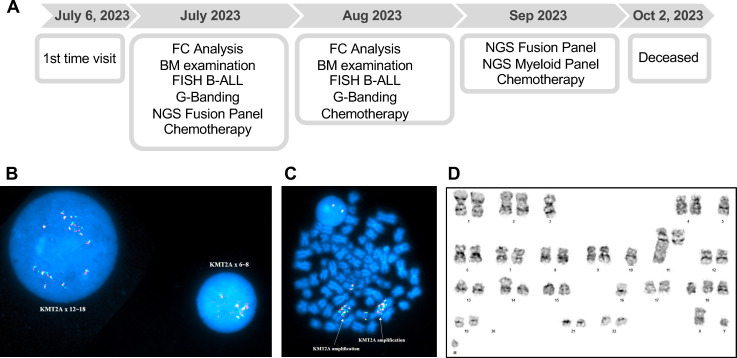 Figure 1
Figure 1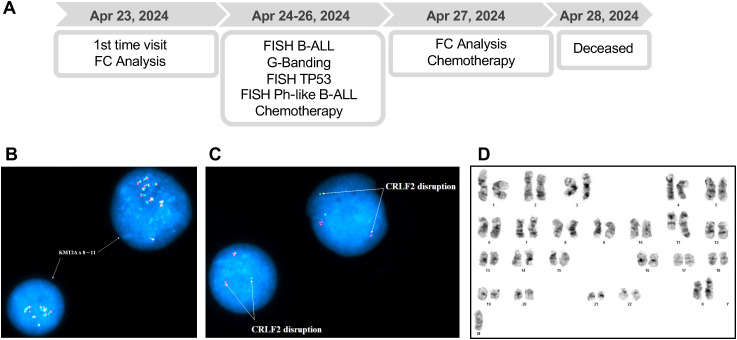 Figure 2
Figure 2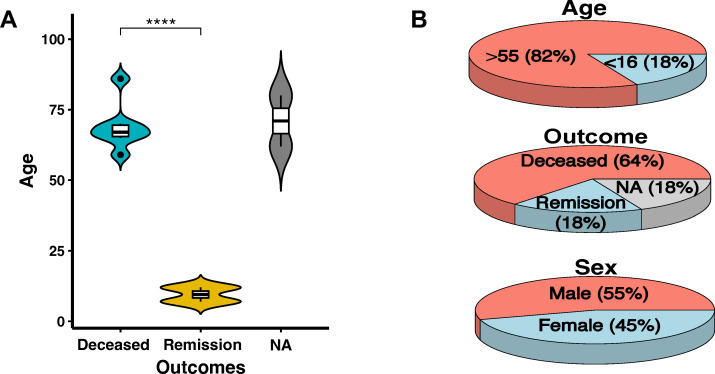 Figure 3
Figure 3| Case | Reference | Sex | Age (Yrs) | Diagnosis | CD10 (FC) | Karyotype |
| Other results | Outcome | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Current case 1 | M | 59 | B-ALL | Neg | 41~44,XY,-3,-5,-10,add(11)(q23),add(14)(p11.2),-16,add(18)(q23),-20,+1~3mar[cp4]/70~80<3n>,XXY,+2,-3,-5,+6,-10,+11,add(11)(q23)x2,+13,+13,add(14)(p11.2),+15,-16,add(16)(q24),add(18)(q23),+21,+22,+2~5mar[cp10] | 6~18 | c.524G>A, p.Arg175His, VAF 91% | 3–5 copies of | Deceased |
| 2 | Current case 2 | F | 66 | Ph-like B-ALL | Pos | Chromosome analysis failed, but metaphases revealed unknown material on chromosome 11q23 | 8~11 | No deletion/duplication | Pos for | Deceased |
| 3 | PMID: 12604431 | F | 86 | pre-B ALL | Neg | 44,XX,del(5)(q13q31),dic(6;17)(p25;q11),hsr(11)(pter_q23::hsr::q21::hsr::qter),-16,-17[18]/46,XX[2] | Multiple | Chr 17 loss | NA | Deceased |
| 4 | PMID: 23238285_1 | M | 80 | Therapy-related B-ALL | Neg | 42,XY,−3,hsr(11)(q23),−14,−16,−20[cp6]/43,idem,+mar[cp3]/46,XY[11] | Multiple | NA | Neg for | NA |
| 5 | PMID: 38735761 | F | 70 | pre-B ALL | Neg | 44-47,XX,del(5)(q22q35),-7,dic(12;18)?(?p12;p11.3),der(15)t(?7;15)(p11.2;p11.2),-20, del(22)(q13.1),+1~4mar[cp6]/44,idem,der(11),add(11)(p15)add(11)(q23)[cp6]/83–87<4n>,XXXX,-2,-3,-4,del(5)(q22q35)x2,-7,-9,-11,add(11)(q?23)x2,-13,-14,-15,-15,-19,-20,-21,+8~10mar[cp4]/46,XX[2] | ≥4 | Biallelic p.Val173Met, p.Val216Met, VAFs 37% and 40%, respectively | NA | Deceased |
| 6 | PMID: 36964033 | M | 69 | B-ALL | Neg | NA | >20 | Deletion | 3–4 copies of | Deceased |
| 7 | Proc UCLA Healthc 19 (2015) | F | 67 | Therapy-related B-ALL | Pos | 45-46,XX,add(8)(q24.3),add(16)(q22),+21,del(21)(q22),-22,add(22)(q11.2),+mar[cp8]/46,XX[8] | 3~4 | NA | Neg for | Deceased |
| 8 | PMID: 35402256 | F | 65 | B-ALL to T-ALL | Neg | 44,X,-X,add(1)(p13),add(2)(q21),-4,-5,-10,del(11)(q)?,-12,-14,-17,-18,+r1,+mar1, +mar2,+mar3,+mar4,+mar5 [3]/46,XX[4] | 8 | c.455dupC, p.Pro153Alafs*28, VAFs 49.5-94.1% | Neg for | Deceased |
| 9 | PMID: 23238285_2 | M | 62 | Therapy-related B-ALL | Neg | 44-45,XY,−5,hsr(11)(q23),−15,+mar1[cp3]/48–50,sl,+6,+8,+20,+22[cp2]/57−59,sdl1,+1,+del(1)(q12),+2,+7,+10,+hsr(11)(q23)a,+12,+13,+21,−mar1,+mar2(cp3)/59,sdl2,+X,−1,−del(1)(q12),+del(1)(q25),+5,+10,+11[cp6]/73–77,sdl2,+Y,+del(1)(q12),+3,+4,+5,+6,+8,+9,+10,+11,−hsr(11),+12,+13,+14,+15,+16,+17,+17,+18,+18,+19,+22,−mar2,+mar3x2[cp7] | Multiple | NA | Neg for | NA |
| 10 | PMID: 22052166 | M | 12 | B-ALL | Pos | 47,X,-Y,+1,del(1)(q25),del(1)(q12),dup(11)(q24q23),add(12)(p11.2),+21 | 3 | NA | NA | Remission |
| 11 | PMID: 11069023 | M | 7 | B-ALL | NA | Failed | >2 | NA | Failed for SB | Remission |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Lymphoblastic Leukemia research · Acute Myeloid Leukemia Research · Chronic Myeloid Leukemia Treatments
Introduction
KMT2A (lysine methyltransferase 2A), formerly known as MLL (mixed-lineage leukemia), is a gene on chromosome band 11q23, which encodes a histone methyltransferase that methylates H3K4, activating key developmental genes and regulating self-renewal and differentiation in hematopoietic stem cells (1). KMT2A rearrangements are frequently observed in both myeloid and lymphoid leukemias, particularly in acute myeloid leukemia (AML) and B-cell acute lymphoblastic leukemia (B-ALL) (2, 3). Unlike KMT2A rearrangements, which involve the fusion of KMT2A with various partner genes, KMT2A amplification arises through mechanisms such as extrachromosomal double minutes or intrachromosomal tandem duplications, often presenting as homogeneously staining regions (HSRs) (4). KMT2A amplification is a rare genetic abnormality, occurring in approximately 1% of AML cases (5–7) and even less frequently in B-ALL, with only a few reported cases in the literature (8, 9).
Despite its rarity in B-ALL, KMT2A amplification has been associated with distinct clinical and genetic features. One notable aspect is its frequent association with TP53 pathogenic variants, including single nucleotide variants or structural variants, further contributing to the aggressive nature of the disease (9). TP53 is a crucial tumor suppressor that regulates genomic stability and apoptosis. TP53 pathogenic variants impair its function, leading to uncontrolled proliferation, therapy resistance, and poor clinical outcomes (10, 11). KMT2A-amplified B-ALL cases with TP53 pathogenic variants are particularly challenging to treat, demonstrating limited responses to standard therapies and a high risk of relapse (9). Given this association, understanding the interplay between KMT2A amplification and TP53 variants is crucial for risk stratification and development of targeted therapies to improve patient outcomes.
CRLF2 rearrangements, a hallmark of Ph-like B-ALL, contribute to cytokine receptor-mediated leukemogenesis by activating the JAK-STAT pathway, leading to enhanced proliferation and poor prognosis (12, 13). About 55–68% of CRLF2-rearranged cases also have JAK-pathway mutations (most often JAK2) (14). CRLF2 rearrangements have not been documented in KMT2A-amplified B-ALL. Recent adult cohort data from a resource-limited setting confirmed that Ph-like B-ALL is common and enriched for adverse features such as MRD positivity and IKZF1 deletions, underscoring its aggressive clinical behavior (15). Since both CRLF2 rearrangements and KMT2A amplification define high-risk B-ALL subtypes and are associated with treatment resistance and poor clinical outcomes, it remains unclear whether they coexist or represent distinct oncogenic mechanisms. If both alterations co-occur, epigenetic dysregulation and JAK-STAT hyperactivation may drive an ultra-high-risk leukemia, requiring combined JAK inhibitors and epigenetic modulators. Further research is needed to assess their coexistence, interactions, and treatment strategies.
Given the rarity of KMT2A amplification in B-ALL, systematic documentation and analysis of cases are essential to improve our understanding of its pathogenesis and refine the diagnostic and therapeutic strategies. In this study, we report two adult B-ALL patients with complex karyotypic abnormalities and KMT2A amplification, including one patient with a TP53 pathogenic variant (p.Arg175His) and another with a CRLF2 rearrangement, marking the first documented case of KMT2A amplification coexisting with Ph-like B-ALL in an adult. Additionally, we review our cases along with nine recently reported cases, providing further insight into the clinical, cytogenetic, and molecular characteristics of KMT2A-amplified B-ALL, and addressing associated management challenges.
Materials and methods
Flow cytometry analysis
Bone marrow specimens from our patients underwent flow cytometry analysis at the University of Alabama at Birmingham (UAB) Pathology Lab. The analysis was conducted following standard flow cytometry protocols to ensure accuracy and reproducibility. The cell markers analyzed by flow cytometry included T-cell markers (CD2, CD3, CD4, CD5, CD7, CD8), B-cell markers (CD9, CD10, CD19, CD20, CD22, Kappa, Lambda), myeloid and progenitor markers (CD13, CD14, CD15, CD16, CD33, CD34, CD38, CD58, CD64, CD117), and the pan-leukocyte marker CD45.
G-banded chromosome and FISH analyses
Bone marrow or peripheral blood specimens from our patients underwent comprehensive cytogenomic analyses, including G-banded chromosome and fluorescence in situ hybridization (FISH) analyses, at the UAB Cytogenetics Lab. G-banded chromosome analysis was performed using standard cytogenetic protocols. Interphase and/or metaphase FISH analyses were conducted following the manufacturer’s protocols, as previously described (16). The B-ALL FISH panel (Abbott) included BCR/ABL1 dual-fusion probes, KMT2A break-apart probes, ETV6/RUNX1 dual-fusion probes, and centromeric probes for chromosome 4, 10, and 17. The Ph-like B-ALL FISH panel (OGT Cytocell) included break-apart probes for ABL2 (1q25.2), PDGFRB (5q32), JAK2 (9p24.1), ABL1 (9q34.1), and CRLF2 (Xp22.33/Yp11.32). The TP53 probe mixture (Abbott) includes a TP53 (17p13.1) probe and a chromosome 17 centromere probe. Findings were reported in accordance with the International System for Human Cytogenomic Nomenclature (ISCN) 2020.
Targeted next-generation sequencing panel analysis
Targeted next-generation sequencing (NGS) panel analysis was conducted on bone marrow biopsy samples at the UAB Pathology Lab to detect gene fusions and somatic variants using an RNA-based myeloid fusion panel and a myeloid mutation panel, following established protocols (17, 18). The RNA-based myeloid fusion panel targeted key fusion driver genes, including ABL1, ABL2, BCL2, BRAF, CCND1, CREBBP, EGFR, ETV6, FGFR1, FGFR2, FUS, HMGA2, JAK2, KAT6A (MOZ), KAT6B, KMT2A, KMT2A-PTDs, MECOM, MET, MLLT10, MRTFA (MKL1), MYBL1, MYH11, NTRK2, NTRK3, NUP214, NUP98, PAX5, PDGFRA, PDGFRB, RARA, RUNX1, TCF3, TFE3, and ZNF384. The myeloid mutation panel assessed hotspot variants in genes such as ABL1, ANKRD26, BRAF, CBL, CSF3R, DDX41, DNMT3A, FLT3, GATA2, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, WT1, MPL, MYD88, NPM1, NRAS, PPM1D, PTPN11, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, and U2AF1, along with full-gene sequencing of ASXL1, BCOR, CALR, CEBPA, ETV6, EZH2, IKZF1, NF1, PHF6, PRPF8, RB1, RUNX1, SH2B3, STAG2, TET2, TP53, and ZRSR2.
Data visualization and statistical analysis
Data visualization and statistical analysis were performed in R (v.4.3.1). A violin plot was generated using the ggviolin function in the ggpubr (v.0.6.0) package in R, while pie charts were created using the pie3D function in the plotrix (v.3.8-6) package. These visualizations illustrate the age distribution, clinical outcomes, and sex distribution of 11 B-ALL patients with KMT2A amplification, with the violin plot representing age distribution and the pie chart depicting clinical outcomes and sex proportions. Tables summarizing patient characteristics and analytical results were generated using the GT (v.0.10.1) package, ensuring clear and structured data presentation. A Student’s t-test was applied for comparisons between deceased and remission groups, with a p-value < 0.05 considered statistically significant.
Case presentations
Patient 1
A 58-year-old male with a history of hypertension (HTN), hyperlipidemia (HLD), gastroesophageal reflux disease (GERD), and anxiety/depression was transferred from an outside hospital for suspected newly diagnosed acute leukemia. He was admitted to UAB for comprehensive diagnostic evaluation. The patient’s clinical course and diagnostic workup are shown in Figure 1A. Flow cytometry of the bone marrow aspirate identified 54.11% leukemic blasts, positive for CD13dim, CD19, CD22dim, CD34, CD38, CD45, CD58, and nTdT, and negative for NK/T, other myeloid, and monocytic markers, confirming a B-ALL diagnosis (Supplementary Table S1). Bone marrow morphology assessment revealed a hypercellular marrow (approximately 90% cellularity) with markedly reduced trilineage hematopoiesis. Most blasts on touch imprint exhibited cytoplasmic vacuolization. These findings are consistent with B-ALL.
(A) Timeline of diagnostic evaluations and clinical course of Patient 1. FC: Flow Cytometry, BM: Bone Marrow, FISH: Fluorescence in situ hybridization, B-ALL: B-cell acute lymphoblastic leukemia panel. (B) Interphase and (C) metaphase FISH analyses of Patient 1 unstimulated bone marrow (BM) cells using the KMT2A BAP, showing KMT2A amplification (618 copies). (D) G-banded chromosome analysis of Patient 1 unstimulated BM cells, demonstrating a stemline clone with complex karyotype involving chromosomes 3, 5, 10, 11, 14, 16, 18, and 20, in addition to the presence of 13 marker chromosomes.
Interphase FISH analysis of uncultured bone marrow cells, using the B-ALL FISH panel, identified 618 copies of KMT2A in 68.5% of cells (Figures 1B, C), along with copy number alterations affecting several genes, including ABL1 (35 copies in 68% of cells), ETV6 (25 copies in 67.5% of cells), RUNX1 (34 copies in 9.5% of cells), and chromosomes 4, 10, and 17 (35 copies each) (Supplementary Table S1). G-banded chromosome analysis of unstimulated bone marrow cells revealed the presence of a stemline clone with complex karyotype involving chromosomes 3, 5, 10, 11, 14, 16, 18, and 20, in addition to the presence of 13 marker chromosomes, and a subclone that represents the inexact doubling product of the stemline clone: 4144,XY,-3,-5,-10,add(11)(q23),add(14)(p11.2),-16,add(18)(q23),-20,+13mar[cp4]/7080<3n>,XXY,+2,-3,-5,+6,-10,+11,add(11)(q23)x2,+13,+13,add(14)(p11.2),+15,-16,add(16)(q24),add(18)(q23),+21,+22,+25mar[cp10] (Figure 1D).
A targeted myeloid NGS panel identified a clinically significant TP53 missense variant (c.524G>A, p.Arg175His) with a 91% variant allele frequency (VAF) (Supplementary Table S1), suggesting a potential role in leukemogenesis and treatment resistance. Additionally, no fusion transcripts were detected using a targeted RNA-based myeloid fusion NGS panel.
The patient was started on induction chemotherapy (Figure 1A), which was complicated by febrile neutropenia, necessitating the removal of his mediport as a potential infection source. During hospitalization, he developed progressive hypoxia, pleural effusions, and encephalopathy, requiring multiple Medical Emergency Team (MET) activations, including one for unresponsiveness, which improved with Narcan. Bronchoscopy showed minimal secretions and arytenoid inflammation, suggesting aspiration. His condition worsened, requiring MET activation for altered mental status and hypotension. He developed severe lactic acidosis (lactate level of 11 mmol/L), tested positive for COVID19, and had persistent pseudomonas and klebsiella bacteremia. Despite maximum vasopressor support, he remained profoundly hypotensive. He ultimately succumbed to refractory septic shock and multiorgan failure after a two-month hospitalization.
Patient 2
A 66-year-old female with a history of type 2 diabetes mellitus, GERD, HTN, obstructive sleep apnea, and HLD presented to an outside hospital with dizziness, ear drainage, gait difficulty, shortness of breath, and cough. Patient’s workup revealed leukocytosis, anemia, and thrombocytopenia, raising suspicion for acute leukemia. She was transferred to UAB for further evaluation. The patient’s diagnostic timeline and treatment course are illustrated in Figure 2A. Flow cytometry of the peripheral blood demonstrated 89.71% leukemic blasts that were positive for CD10, CD13, CD19, CD22dim, CD34, CD38, CD58, nTdT, and negative for NK/T, other myeloid, and monocytic cell markers, supporting a diagnosis of B-ALL (Supplementary Table S1).
(A) Timeline of diagnostic evaluations and clinical course of Patient 2. FC: Flow Cytometry, FISH: Fluorescence in situ hybridization, B-ALL: B-cell acute lymphoblastic leukemia panel. (B) Interphase FISH analysis of Patient 2 peripheral blood cells using the KMT2A BAP, showing KMT2A amplification (8~11 copies). (C) Interphase FISH analysis of Patient 2 unstimulated BM cells using the CRLF2 BAP, demonstrating CRLF2 rearrangement. (D) G-banded chromosome analysis of Patient 2 peripheral blood cells were unsuccessful; however, limited analysis revealed additional material of unknown origin attached to 11q23.
Interphase FISH analysis using the B-ALL FISH panel identified KMT2A amplification, with 8~11 copies detected in 87.5% of peripheral blood cells (Figure 2B). Additionally, the Ph-like B-ALL FISH panel analysis detected a CRLF2 gene rearrangement in 85% of bone marrow cells (Figure 2C), with the classic fusion signal disruption suggestive of the t(X;14)(p22.3;q32.3) and resulting in IGH::CRLF2 fusion. This is an alteration commonly associated with high-risk leukemia and poor treatment response. Further FISH analysis of the TP53 gene in bone marrow cells showed no deletions or structural rearrangements (Supplementary Table S1). G-banded chromosome analysis of peripheral blood cells was unsuccessful, but limited analysis indicated additional material attached to chromosome 11 at band 11q23 (Figure 2D), a region frequently involved in KMT2A-related leukemias.
During hospitalization, the patient developed pulmonary edema and decompensated cirrhosis, accompanied by altered mental status and respiratory distress, requiring MET activation, Bilevel Positive Airway Pressure support, and eventual intubation. Despite treatment with Lasix, Solumedrol, broad-spectrum antibiotics, and blood transfusions, her condition worsened due to septic shock, acute kidney injury requiring continuous renal replacement therapy, non-ST elevation myocardial infarction, and cirrhosis with varices. Given her multiorgan failure and poor prognosis, goals of care discussions led to a comfort care transition. She was later found unresponsive with no pulse, and electrocardiogram confirmed no cardiac rhythm. She passed away four days after hospitalization (Figure 2A). Due to her rapid clinical deterioration and subsequent death, targeted NGS could not be performed.
Cases review
This report reviews two cases from our cohort and nine published cases of KMT2A-amplified B-ALL (Table 1), summarizing distributions by age (9/11, 82% >55 years; 2/11, 18% <16 years), outcome (7/11, 64% deceased; 2/11, 18% in remission; 2/11, 18% not available), and sex (6/11, 55% male; 5/11, 45% female). Both pediatric patients (6, 19) (18%) achieved remission, whereas the majority of adult patients (64%) experienced fatal outcomes, suggesting a strong age-associated difference in clinical course. Patients in the remission group clustered at markedly younger ages than those in the deceased group, and fatal cases were predominantly observed among older adults (Figures 3A, B). Among the eleven cases analyzed, one case (9%) was classified as Ph-like B-ALL, two cases (18%) as pre-B ALL (9, 20), three cases (27%) as therapy-related B-ALL (21, 22), and one case (9%) exhibited transformation from B-ALL to T-ALL (23). CD10 expression varied, with three cases positive (27%) and seven negative (63%), indicating immunophenotypic diversity. KMT2A amplification (3 to >20 copies) was frequently associated with complex karyotypes, particularly involving chromosome band 11q23. TP53 alterations were found in five cases (45%), including missense variants or structural variants. Additional genetic changes included CRLF2 rearrangement (1/11, 9%), multiple copies of ABL1, BCR, ETV6, and RUNX1, as well as chromosomal deletions. These findings highlight the need for further research and novel therapies for this high-risk leukemia subtype.
*(A) Violin plot showing age and outcome distribution of B-ALL patients with KMT2A amplification (our two patients and nine published cases). Outcomes: Deceased (blue), Remission (yellow), NA (not available) (gray). ***p-value < 0.001. (B) Pie charts summarizing age (82% >55 years, 18% <16 years), outcome (64% deceased, 18% remission, 18% NA), and sex (55% male, 45% female) distribution.
Discussion
KMT2A amplification is a rare genetic abnormality in B-ALL, with few documented cases (Table 1). The aggregated cases summarized in Table 1 indicate that KMT2A-amplified B-ALL predominantly affects older adults, is frequently associated with complex karyotypes involving chromosome band 11q23, a high rate of TP53-related alterations including TP53 variants in three cases and loss of chromosome 17 in one case (45%), CD10 negativity (63%), and carries a poor overall prognosis, with nearly two-thirds of reported patients deceased. Our findings further confirm its strong association with TP53 pathogenic variants. Patient 1 exhibited a clinically significant TP53 missense variant (c.524G>A, p.Arg175His, VAF 91%), a known driver of genomic instability and poor prognosis in hematologic malignancies. This variant disrupts DNA binding, impairing tumor suppression, and promoting genomic instability, therapy resistance, and cancer progression. Additionally, this variant exhibits oncogenic properties, enhancing proliferation, invasion, metabolic reprogramming, and angiogenesis (24). TP53 single nucleotide variants or structural variants were also identified in five previously reported cases, supporting its role as a cooperative oncogenic event in KMT2A-amplified B-ALL. Case 3 exhibited a chromosome 17 loss (20), while case 5 had biallelic TP53 variants (p.Val173Met and p.Val216Met, VAFs 37% and 40%, respectively) (9), suggesting significant TP53 loss. Case 6 showed a TP53 deletion (8), reinforcing the link between TP53 loss and genomic instability, while case 8 carried a TP53 frameshift variant (c.455dupC, p.Pro153Alafs*28, VAFs 49.5–94.1%) (23). Given the frequent co-occurrence of TP53 pathogenic variants and KMT2A amplification, future studies should explore TP53-targeted therapies for this high-risk B-ALL subset. Prior work in therapy-related leukemia has reported an association between KMT2A copy-number gain and germline TP53 alterations (25). However, germline testing was not performed for our patients due to lack of an appropriate non-tumor specimen, and family history was non-contributory based on available documentation. Notably, a very high variant allele fraction (VAF) of 91% was observed for the TP53 variant in Patient 1, which may reflect a homozygous TP53 variant due to copy-neutral loss of heterozygosity (LOH) or a hemizygous TP53 variant resulting from a deletion of the other allele involving this region. However, in the absence of germline testing or detailed family history, it is uncertain what underlies the high allele frequency for this TP53 variant and whether this variant is germline. Future studies should systematically evaluate germline TP53 status in this rare subgroup to clarify inherited versus acquired risk.
KMT2A abnormalities in B-ALL are most described as KMT2A rearrangements, which are frequent in infant ALL. In contrast, KMT2A amplification is a mechanistically distinct alteration involving copy-number gain rather than gene fusion. Although KMT2A rearrangements have been reported in Ph-like ALL, KMT2A amplification has not previously been described in adult Ph-like B-ALL, to the best of our knowledge. Increased KMT2A dosage may lead to partial functional convergence with rearranged cases through dysregulation of shared transcriptional programs such as the HOXA/MEIS1 axis. In this context, this case report identifies the first documented co-occurrence of KMT2A amplification and CRLF2 rearrangement in an adult with de novo Ph-like B-ALL. However, targeted NGS could not be performed for Patient 2 due to rapid clinical deterioration, representing a limitation of this study and an important area for future investigation. CRLF2 rearrangements, a hallmark of Ph-like B-ALL, result in either IGH::CRLF2 or P2RY8::CRLF2 fusion with CRLF2 overexpression, which drive JAK-STAT activation, and are associated with poor treatment response and high relapse rates (12, 13). While well-studied in Ph-like B-ALL, their co-presence in KMT2A-amplified cases remains unreported. Although clonal co-localization could not be directly demonstrated, the high percentage of both abnormalities and the presence of a single dominant blast population strongly support their occurrence within the same leukemic clone, suggesting particularly aggressive disease biology.
Recognition of KMT2A amplification, especially with TP53 alterations and/or CRLF2 rearrangement, has management implications beyond prognosis and should prompt early, genomically informed escalation of care. Menin inhibitors (revumenib, ziftomenib) represent a rational strategy to disrupt KMT2A-associated transcriptional programs (26, 27), while JAK inhibition (ruxolitinib) is supported for CRLF2-rearranged Ph-like B-ALL (28). Combined pathway targeting may be considered in dual-positive cases. Given poor outcomes with conventional chemotherapy, early clinical trial referral is warranted, and immunotherapies (blinatumomab, inotuzumab, CAR-T) may serve as bridge or salvage options (29–31).
Cytogenetic complexity is another key feature of KMT2A-amplified B-ALL. Both patients exhibited complex karyotypes with multiple chromosomal abnormalities, particularly involving chromosome band 11q23, consistent with previous reports linking KMT2A amplification to genomic instability. Additional copy number abnormalities in genes such as ABL1, BCR, ETV6, and RUNX1 suggest that secondary genetic alterations may further drive disease aggressiveness and therapeutic resistance.
Our review of 11 KMT2A-amplified B-ALL cases (two from our cohort and nine published) highlights key clinical trends. Most patients (82%) were older adults (median age 66), with a poor prognosis, as 81.8% succumbed to the disease. The two survivors were pediatric cases, suggesting age may influence prognosis due to differences in disease biology or treatment response. The observed outcome difference between the deceased and remission groups likely reflects an age-associated difference in clinical course, with pediatric cases clustering in the remission group and older adults predominating among fatal cases. However, this observation is presented for exploratory and descriptive purposes only, as formal statistical comparison is limited by marked age imbalance and small sample size, which limit adjustment for confounders. The CD10 negativity (63%) was common, aligning with the aggressive nature of KMT2A-rearranged B-ALL. Pre-B ALL accounted for 18% of cases, therapy-related B-ALL for 27%, and B-ALL to T-ALL transformation for 9%, with one case of Ph-like B-ALL further expanding the spectrum of high-risk genetic alterations. Given the poor survival and limited treatment options, novel therapeutic strategies are urgently needed. Standard chemotherapy appears ineffective, as shown by rapid disease progression and high mortality. The co-occurrence of KMT2A amplification in this B-ALL entity with TP53 pathogenic variants or CRLF2 rearrangement suggests that targeted therapies, including p53 reactivators, JAK inhibitors, and/or epigenetic modulators, should be explored. The potential role of immunotherapy, such as CAR-T cells or bispecific antibodies, also warrants further investigation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Castiglioni S Di Fede E Bernardelli C Lettieri A Parodi C Grazioli P . KMT 2A: umbrella gene for multiple diseases. Genes (Basel). (2022) 13:514. doi: 10.3390/genes 13030514, PMID: 35328068 PMC 8949091 · doi ↗ · pubmed ↗
- 2Gorecki M Koziol I Kopystecka A Budzynska J Zawitkowska J Lejman M . Updates in KMT 2A gene rearrangement in pediatric acute lymphoblastic leukemia. Biomedicines. (2023) 11:746. doi: 10.3390/biomedicines 11030821, PMID: 36979800 PMC 10045821 · doi ↗ · pubmed ↗
- 3Hernandez-Sanchez A Gonzalez T Sobas M Strang E Castellani G Abaigar M . Rearrangements involving 11q 23.3/KMT 2A in adult AML: mutational landscape and prognostic implications - a HARMONY study. Leukemia. (2024) 38:1929–37. doi: 10.1038/s 41375-024-02333-4, PMID: 38965370 PMC 11347382 · doi ↗ · pubmed ↗
- 4Hess JL . Mechanisms of transformation by MLL. Crit Rev Eukaryot Gene Expr. (2004) 14:235–54. doi: 10.1615/Crit Rev Eukaryot Gene Expr.v 14.i 4.10, PMID: 15663355 · doi ↗ · pubmed ↗
- 5Tang G Di Nardo C Zhang L Ravandi F Khoury JD Huh YO . MLL gene amplification in acute myeloid leukemia and myelodysplastic syndromes is associated with characteristic clinicopathological findings and TP 53 gene mutation. Hum Pathol. (2015) 46:65–73. doi: 10.1016/j.humpath.2014.09.008, PMID: 25387813 · doi ↗ · pubmed ↗
- 6Cuthbert G Thompson K Mc Cullough S Watmore A Dickinson H Telford N . MLL amplification in acute leukaemia: a United Kingdom Cancer Cytogenetics Group (UKCCG) study. Leukemia. (2000) 14:1885–91. doi: 10.1038/sj.leu.2401919, PMID: 11069023 · doi ↗ · pubmed ↗
- 7Andersen MK Christiansen DH Kirchhoff M Pedersen-Bjergaard J . Duplication or amplification of chromosome band 11q 23, including the unrearranged MLL gene, is a recurrent abnormality in therapy-related MDS and AML, and is closely related to mutation of the TP 53 gene and to previous therapy with alkylating agents. Genes Chromosomes Cancer. (2001) 31:33–41. doi: 10.1002/gcc.1115, PMID: 11284033 · doi ↗ · pubmed ↗
- 8Wren C Rebeiro P Tegg E . KMT 2A amplification in B lymphoblastic leukaemia. Pathology. (2023) 55:738–40. doi: 10.1016/j.pathol.2022.12.356, PMID: 36964033 · doi ↗ · pubmed ↗