Case Report: Somatic NF2 mutation in a vestibular schwannoma arising in a patient with neurofibromatosis type 1
Misa Shogaku, Hiroshi Yamada, Seiji Yamada, Ryota Fujinami, Shigeki Yamada, Motoki Tanikawa, Yusuke Okuno, Mitsuhito Mase

TL;DR
A rare case of a vestibular schwannoma in a patient with neurofibromatosis type 1 is linked to a somatic NF2 mutation and chromosome 22 loss.
Contribution
This case report identifies a somatic NF2 mutation as a driver of vestibular schwannoma in an NF1 patient.
Findings
The patient had a germline NF1 mutation present in all tissues analyzed.
The vestibular schwannoma had a somatic NF2 mutation and loss of chromosome 22.
The NF2 mutation and chromosome 22 loss were absent in the neurofibroma and blood samples.
Abstract
Neurofibromatosis type 1 (NF1) and vestibular schwannoma are genetically and clinically distinct entities, with vestibular schwannomas classically associated with neurofibromatosis type 2. The occurrence of a vestibular schwannoma in a patient with NF1 is rare, and its underlying molecular mechanism remains unclear. We report a 51-year-old man clinically diagnosed with neurofibromatosis type 1 who developed a unilateral vestibular schwannoma presenting with progressive hearing loss and neurological symptoms. Histopathological examination following surgical resection confirmed the diagnosis of vestibular schwannoma, and genetic analyses were subsequently performed on the schwannoma, a cutaneous neurofibroma, and peripheral blood from the same patient. Whole-exome sequencing revealed a pathogenic germline NF1 mutation shared across all analyzed samples. In contrast, the vestibular…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
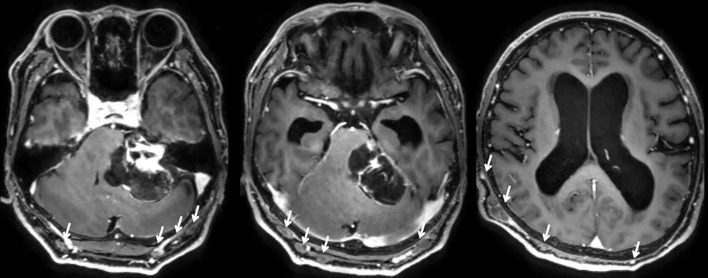 Figure 1
Figure 1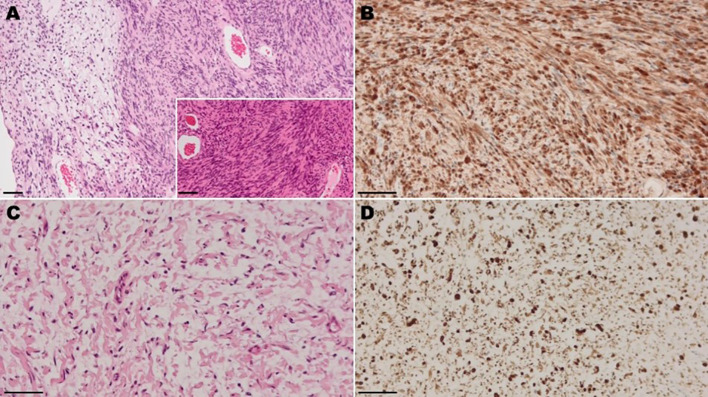 Figure 2
Figure 2| Tissue | Mutation | Zygosity |
|---|---|---|
| Peripheral blood | Heterozygous | |
| Neurofibroma | Heterozygous | |
| Vestibular schwannoma | Heterozygous | |
| Vestibular schwannoma | Heterozygous | |
| Vestibular schwannoma | Heterozygous | |
| Vestibular schwannoma | Heterozygous | |
| Vestibular schwannoma | Heterozygous | |
| Vestibular schwannoma | Hemizygous | |
| Vestibular schwannoma | Heterozygous | |
| Vestibular schwannoma | Heterozygous | |
| Vestibular schwannoma | chr22 loss | Heterozygous |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurofibromatosis and Schwannoma Cases · Meningioma and schwannoma management · Soft tissue tumor case studies
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominant neurocutaneous disorder characterized by a range of clinical manifestations, including multiple neurofibromas, café-au-lait macules, inguinal freckling, optic gliomas, Lisch nodules, and skeletal abnormalities. The NF1 gene, located on chromosome 17q11.2, encodes the protein neurofibromin. Loss of neurofibromin leads to hyperactivation of the Ras/MAPK and mechanistic target of rapamycin (mTOR) signaling pathways, thereby contributing to tumorigenesis (1). Approximately 50% of NF1 cases are inherited, while the remainder arise de novo, with no family history (2). Clinical presentations vary widely and are likely influenced by epigenetic modifications (1). Neurofibromas develop in about 60% of NF1 patients and may occur either cutaneously or internally. Plexiform neurofibromas, which are pathognomonic for NF1, arise internally and may undergo malignant transformation into peripheral nerve sheath tumors (MPNSTs), with a lifetime risk of estimated at 8–13% (1).
Neurofibromatosis type 2 (NF2) is another autosomal dominant disorder, characterized by vestibular schwannomas, spinal schwannomas, meningiomas, and ependymomas, and juvenile cataracts. Vestibular schwannomas are typically solitary tumors; however, approximately 4% to 6% are associated with NF2 (3). The NF2 gene, located on chromosome 22q12.2, encodes the tumor suppressor protein merlin, which influences several key signaling pathways, including PI3 kinase(PI3K)/Akt, Raf/MEK/ERK, and mTOR. Mutations in the merlin gene are identified in approximately 93% of patients with clinical evidence of NF2 and a positive family history. However, more than half of the NF2 cases occur de novo in individuals without any family history (1).
By contrast, NF1 and NF2 are distinct autosomal dominant disorders characterized by different clinical and genetic features. NF1 is primarily associated with cutaneous neurofibromas and Ras pathway dysregulation, while NF2 is typified by vestibular schwannomas due to merlin deficiency. Although historically grouped under the umbrella of “neurofibromatosis,” these two conditions rarely present concurrently in a single patient.
The coincidental occurrence of NF1 and NF2-related manifestations raises intriguing questions about potential genetic crosstalk or sequential mutational events that remain poorly characterized. To date, reports of such overlap remain exceedingly rare.
Here, we describe a unique case of a patient fulfilling clinical criteria for NF1 who concurrently developed a vestibular schwannoma, with genetic analysis revealing both a germline NF1 mutation and a somatic NF2 mutation in tumor tissue.
Case presentation
A 51-year-old man presented with left-sided hearing loss and dysarthria. He also reported a progressive gait disturbance that had gradually worsened over several months. Neurological examination revealed left-sided sensorineural hearing loss (4-frequency pure-tone average of 62.5 dB), facial nerve palsy, and dysmetria with associated coordination disturbances.
The patient had a long-standing history of cutaneous abnormalities, including multiple pigmented macules and subcutaneous nodules, which had not previously been evaluated. Dermatological consultation identified these lesions as café-au-lait spots and neurofibromas, leading to a clinical diagnosis of NF1. The patient also reported a family history of similar skin findings in his father, although no formal diagnosis or treatment had been documented. Other diagnostic criteria for NF1, such as axillary freckling, Lisch nodules, or skeletal dysplasia, were absent.
Contrast-enhanced magnetic resonance imaging (MRI) of the head revealed a heterogeneously enhancing mass, approximately 5 cm in diameter, located in the left cerebellopontine (CP) angle (Figure 1). The lesion exerted significant compression on the cerebellum and brainstem, caused enlargement of the internal auditory canal, and led to obstructive hydrocephalus. The tumor exhibited both solid components with intense gadolinium enhancement and cystic areas. Notably, multiple subcutaneous masses were also visualized on the same imaging studies.
Preoperative axial contrast enhanced T1-weighted MRI demonstrating a mass lesion in the left cerebellopontine angle. The lesion shows well-enhanced solid components and cystic components, causing significant compression of the cerebellum and brain stem, along with associated hydrocephalus. Additionally, multiple subcutaneous mass lesions are observed (white arrows).
The CP angle mass was subtotally resected via a lateral suboccipital craniotomy. A subcutaneous mass lesion located near the surgical incision was also excised. The postoperative course was uneventful, with improvement in neurological symptoms except for complete deafness on the operated side, and follow-up MRI has shown no evidence of tumor regrowth to date. Histopathological examination confirmed the CP angle lesion as a schwannoma, showing characteristic Antoni A and B patterns (Figure 2A), with diffuse S-100 positivity (Figure 2B). The schwannoma showed a low Ki-67 labeling index of approximately 1%. In contrast, the subcutaneous tumor was consistent with a neurofibroma, showing thin wavy cells in a myxoid stroma (Figure 2C) and diffuse S-100 positivity (Figure 2D).
Light micrographs of the surgical specimen (A, B) Intracranial tumor. (A) Hematoxylin and eosin (H&E) staining. The tumor shows biphasic pattern with compact area (Antoni A tissue; right) adjacent to loosely arranged area (Antoni B tissue; left). The tumor of Antoni A tissue is composed of spindle cells showing occasional nuclear palisading, known as Verocay bodies (inset). (B) Immunohistochemically, the tumor cells are diffusely positive for S-100 protein. (C, D) Subcutaneous tumor. (C) H&E staining. The tumor shows proliferation of thin wavy cells immersing in a myxoid stroma. (D) Immunohistochemically, the tumor cells are diffusely positive for S-100 protein.
Methods
Patient
The patient provided informed consent, and the study was approved by the Institutional Review Board (IRB) of Nagoya City University (IRB number: 70-19-0003).
To investigate the underlying genetic etiology, we performed whole-exome sequencing on peripheral blood leukocytes, a subcutaneous neurofibroma, and a vestibular schwannoma, essentially as previously described (4).
Exome sequencing
Briefly, genomic DNA was extracted from fresh tumor tissues and blood samples using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany), following the manufacturer’s instructions. Exome capture was performed using Agilent SureSelect Human All Exon v6, and sequencing was conducted on the Illumina NovaSeq platform (Illumina, San Diego, CA, USA). Germline and somatic point mutations, using the peripheral blood as the reference, were identified using VarScan2 (5) and annotated with ANNOVAR (6). Copy number alterations were detected by comparing sequencing coverage between the sample of interest and 12 unrelated germline samples. Loss of heterozygosity and uniparental disomy were evaluated based on the variant allele frequencies of common single nucleotide polymorphisms (SNP) (minor allele frequency >1%) to identify runs of homozygosity (7).
Results
Our whole-exome sequencing analysis of peripheral blood, a subcutaneous neurofibroma, and a vestibular schwannoma identified a germline NF1 mutation in all three samples, as well as eight somatic genetic alterations in the schwannoma sample (Table 1).
The germline NF1 mutation was a missense variant, c.2351G>T (p.Trp784Leu), which is not listed in either SNP databases or pathogenic mutation databases. However, the 784th amino acid residue is evolutionarily conserved and located within a functionally important domain of neurofibromin, where pathogenic variants have been frequently reported (8, 9), and we interpreted this variant as pathogenic in accordance with the ACMG guidelines (criteria:PM1, PM2, and PP4) (10).
Somatic alterations in the schwannoma included an NF2 frameshift mutation (c.1282delC, p.Gln428Argfs*11) and a deletion of the entire chromosome 22, resulting in the loss of the wild-type NF2 allele. Consequently, the NF2 frameshift mutation was present in a hemizygous state. The remaining six somatic point mutations were considered passenger mutations, as they were not found in somatic mutation databases such as COSMIC (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). No copy number loss or uniparental disomy events affecting NF1 were detected in the vestibular schwannoma.
No somatic mutations were identified in the subcutaneous neurofibroma.
Discussion
The development of schwannomas is a hallmark of NF2, a genetically distinct disorder from NF1. Schwannomas are rare in NF1, with only a limited number of cases reported in the literature (2, 3, 11–17). Nevertheless, patients with NF1 are predisposed to a broad spectrum of tumors, as demonstrated by previous large population-based cohort studies (18). Non-malignant tumors commonly associated with NF1 include neurofibromas, Lisch nodules, optic pathway gliomas, and other low-grade central nervous system gliomas. Malignant neoplasms include malignant peripheral nerve sheath tumors (MPNSTs), juvenile myelomonocytic leukemia (JMML), pheochromocytoma, rhabdomyosarcoma, and glioblastoma multiforme (19). However, vestibular schwannomas in patients with NF1 are exceedingly rare. To the best of our knowledge, the only previously reported case involving genetic analysis was by A. Huq et al., who described a unilateral vestibular schwannoma harboring a somatic NF2 mutation—findings that align with those observed in the present case (2).
Most unilateral vestibular schwannomas arise sporadically and are not associated with germline NF2 mutations (20). However, somatic NF2 mutations have been frequently identified in sporadic vestibular schwannomas, with reported prevalence rates ranging from 15% to 100% (21–28). Furthermore, approximately 50% of NF2 cases are de novo, and over 30% exhibit somatic mosaicism (29, 30). Mohyuddin et al. suggested that the presence of a unilateral vestibular schwannoma in a young individual should prompt evaluation for mosaic NF2 mutations (31). In previously reported NF1 cases with co-occurring schwannomas that underwent genetic analysis, NF2 mutations were identified in the tumor tissue but not in peripheral blood samples, suggesting a somatic origin. However, the possibility of mosaicism could not be completely excluded (2, 15). Yoo et al. described a unique case in which a patient with a germline NF1 mutation exhibited a clinical phenotype more consistent with NF2 than with NF1 (15). They hypothesized that an NF1 splicing mutation might influence tumor formation in tissues harboring NF2 mutations, although the precise mechanism remains unclear.
Taken together, these observations raise the question of why schwannomas, typically associated with NF2, occasionally occur in patients with NF1.To explore this, it is important to consider the biological consequences of NF1 loss at the cellular level.
In patients with NF1, a pathogenic variant is present in one allele of the NF1 gene in all somatic cells, resulting in a baseline susceptibility to tumor development. When the remaining wild-type allele is lost, there is a complete loss of neurofibromin function, which further enhances tumorigenic potential. Neurofibromin negatively regulates the Ras signaling pathway by accelerating the conversion of active Ras-GTP to inactive Ras-GDP. Loss of neurofibromin leads to elevated Ras-GTP levels, which in turn enhance signaling through Raf kinase and activate the downstream MEK and MAPK (Erk1/2) pathways, ultimately promoting cell proliferation (32).
One possible explanation is that germline NF1 mutations may predispose cells to acquiring somatic NF2 mutations. NF1 mutations drive Ras pathway hyperactivation, creating a cellular environment characterized by excessive proliferative signaling and increased genomic instability, consistent with previous observations of elevated Ras-GTP levels and the requirement for additional genetic events in NF1-deficient cells (33).
In the present case, genetic analyses of peripheral blood, vestibular schwannoma, and a subcutaneous neurofibroma revealed a germline NF1 mutation and a somatic NF2 mutation restricted to the schwannoma. These findings support a dual-pathogenic mechanism involving both NF1 and NF2. A permissive tumorigenic background associated with a germline NF1 mutation, together with a somatic NF2 mutation in Schwann cells, may provide a plausible biological explanation for schwannoma development. The possibility of mosaicism cannot be entirely excluded; nevertheless, it was considered unlikely given the unilateral vestibular schwannoma, absence of other NF2-associated features, and negative family history. Ongoing clinical follow-up is warranted to monitor for the potential development of bilateral vestibular schwannomas or other NF2-associated tumors.
Conclusion
This case demonstrates that vestibular schwannoma arising in a patient with neurofibromatosis type 1 can be associated with secondary somatic NF2 alterations accompanied by loss of chromosome 22. By performing comprehensive genetic analyses of three distinct tissues—peripheral blood, a cutaneous neurofibroma, and the vestibular schwannoma—from the same patient, we were able to directly distinguish germline and tumor-specific genetic events, revealing that NF2 alterations were restricted to the vestibular schwannoma. These findings highlight the importance of tumor-specific somatic events, in addition to the germline background, in the development of NF2-related tumors in NF1 patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kresak JL Walsh M . Neurofibromatosis: A review of NF 1, NF 2, and schwannomatosis. J Pediatr Genet. (2016) 5:98–104. doi: 10.1055/s-0036-1579766, PMID: 27617150 PMC 4918700 · doi ↗ · pubmed ↗
- 2Huq A Kentwell M Tirimacco A Rossini J Rawlings L Winship I . Vestibular schwannoma in a patient with neurofibromatosis type 1: clinical report and literature review. Fam Cancer. (2015) 14:157–60. doi: 10.1007/s 10689-014-9763-y, PMID: 25362541 · doi ↗ · pubmed ↗
- 3Araki Y Imai S Funakoshi T Sakai N Yamada H Shimokawa K . A family of von Recklinghausen’s disease associated with bilateral acoustic neurinomas and falx meningiomas. No To Shinkei. (1984) 36:641–8. doi: 10.11477/mf.1406205344 6435649 · doi ↗ · pubmed ↗
- 4Okuno Y Murata T Sato Y Muramatsu H Ito Y Watanabe T . Publisher Correction: Defective Epstein-Barr virus in chronic active infection and haematological Malignancy. Nat Microbiol. (2019) 4:544. doi: 10.1038/s 41564-019-0387-8, PMID: 30705423 · doi ↗ · pubmed ↗
- 5Koboldt DC Zhang Q Larson DE Shen D Mc Lellan MD Lin L . Var Scan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. (2012) 22:568–76. doi: 10.1101/gr.129684.111, PMID: 22300766 PMC 3290792 · doi ↗ · pubmed ↗
- 6Wang K Li M Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e 164. doi: 10.1093/nar/gkq 603, PMID: 20601685 PMC 2938201 · doi ↗ · pubmed ↗
- 7Al-Kzayer LFY Saeed RM Ghali HH Tanaka M Al-Jadiry MF Faraj SA . Comprehensive genetic analyses of childhood acute leukemia in Iraq using next-generation sequencing. Transl Pediatr. (2023) 12:827–44. doi: 10.21037/tp-22-512, PMID: 37305720 PMC 10248931 · doi ↗ · pubmed ↗
- 8Wang W Qin W Ge H Kong X Xie C Tang Y . Clinical and molecular characteristics of thirty NF 1 variants in Chinese patients with neurofibromatosis type 1. Mol Biol Rep. (2019) 46:4349–59. doi: 10.1007/s 11033-019-04888-3, PMID: 31201679 · doi ↗ · pubmed ↗