Investigating the therapeutic profile of velaglucerase alfa in paediatric patients with Gaucher disease: a systematic review across all paediatric age groups
Javier de las Heras, Jorge J Cebolla, Sofía de Pedro, Manuel Gómez-Barrera, Isidro Vitoria

TL;DR
This study reviews the safety and effectiveness of velaglucerase alfa in treating children with Gaucher disease, showing it is well-tolerated and beneficial for most symptoms.
Contribution
The study provides a comprehensive systematic review of velaglucerase alfa's therapeutic profile in all pediatric age groups with Gaucher disease.
Findings
Velaglucerase alfa is well tolerated and effective in treating hematological, visceral, and skeletal symptoms in pediatric GD1 patients.
The treatment maintains stability in patients previously treated with imiglucerase and improves quality of life when administered at home.
For GD3, the drug shows a favorable safety profile and some improvement in non-neurological symptoms, though evidence is limited.
Abstract
Gaucher disease (GD) is a rare autosomal recessive genetic disorder. The clinical manifestations can be adequately managed with enzyme replacement therapy (ERT). The aim of this systematic literature review was to explore the safety and efficacy or effectiveness (depending on the type of evidence) profile of velaglucerase alfa in the treatment of paediatric patients with type 1 (GD1) and type 3 (GD3) GD across all paediatric ages. A systematic review of the PubMed/Medline and Embase databases, along with communications from international conferences, was conducted. The inclusion criteria comprised clinical studies published in either English or Spanish that assessed the therapeutic profile of velaglucerase alfa in patients with GD1 (primarily) and GD3 (exploratorily) of all paediatric ages (0–18 years). For each of the selected publications, data regarding the safety and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
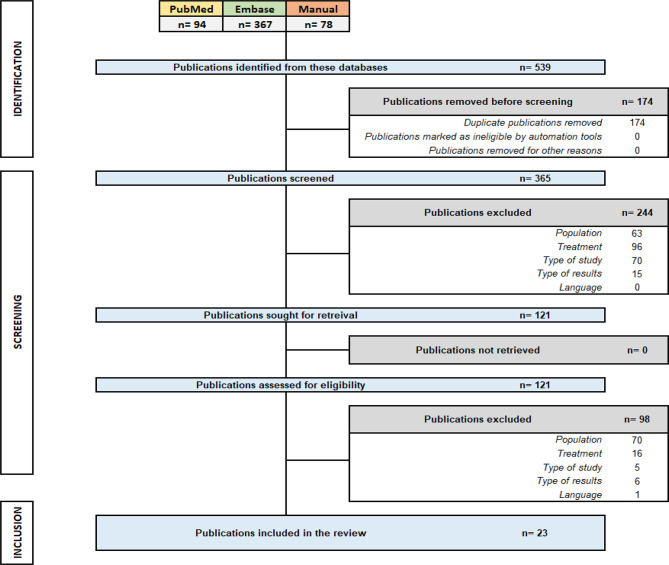 Figure 1
Figure 1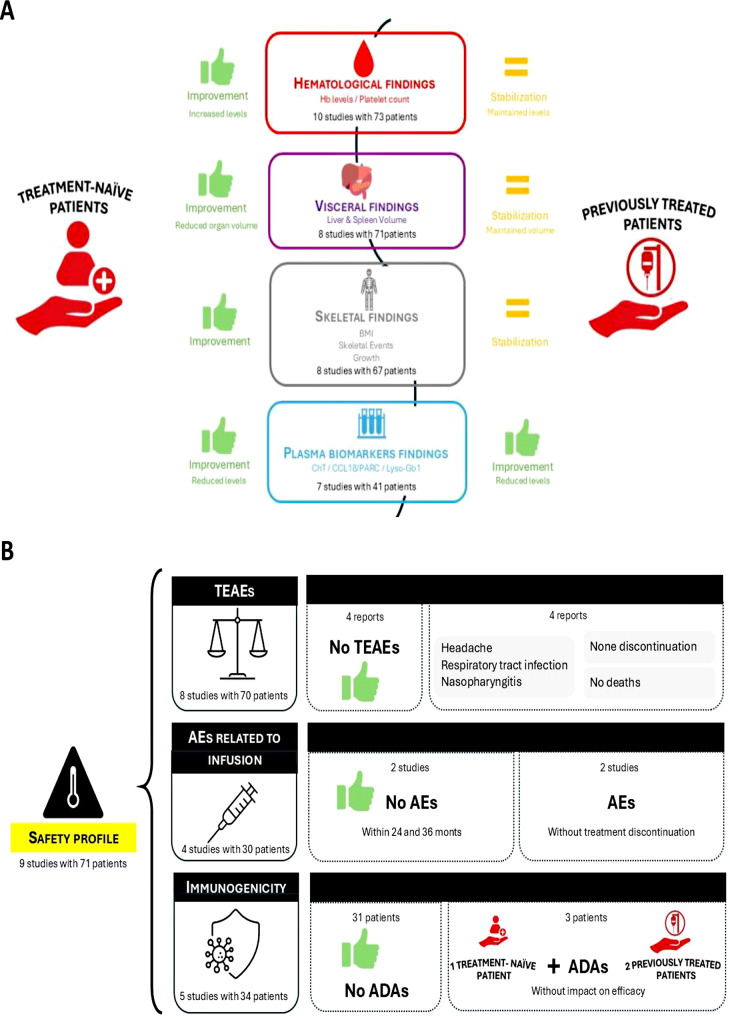 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Glycogen Storage Diseases and Myoclonus · Biochemical and Structural Characterization