Acquired antimicrobial resistance genes in Bordetella Species: a global genomic analysis
Biao Tang, Xiaohe Hu, Yu Song, Xu Liu, Guoping Zhao, Min Yue

TL;DR
This study analyzes antimicrobial resistance genes in non-B. pertussis Bordetella species using global genomic data to understand resistance patterns and transmission.
Contribution
The study provides the first comprehensive global genomic analysis of acquired antimicrobial resistance genes in non-B. pertussis Bordetella species.
Findings
105 antimicrobial resistance genes were identified across 746 genomes of three non-B. pertussis Bordetella species.
B. bronchiseptica showed the highest diversity of resistance genes and multidrug-resistant isolates in lineage 2.
International and cross-host transmission events were detected, including human-dog/rabbit transfers and spread between France and the US.
Abstract
The genus Bordetella comprises Gram-negative pathogens, notably B. pertussis, which causes whooping cough, underscoring the need to characterize antimicrobial resistance (AMR) within this group. However, reports of acquired antimicrobial resistance genes (ARGs) in non-B. pertussis Bordetella species remain scarce. Non–B. pertussis Bordetella genomes were retrieved from NCBI, adapters trimmed and low-quality reads filtered, then assembled for analysis. ARGs were identified with Abricate, ResFinder, and BLAST, and their genomic contexts visualized using Easyfig. Core-genome SNP phylogeny and Bayesian clustering delineated lineages, and SNP-based thresholds quantified transmission. Across 746 genomes, 105 ARGs were identified in three non-B. pertussis species: B. bronchiseptica (96 ARGs), B. trematum (7 ARGs) and B. avium (2 ARGs). Ten distinct ARG types covering sulphonamide (sul1,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
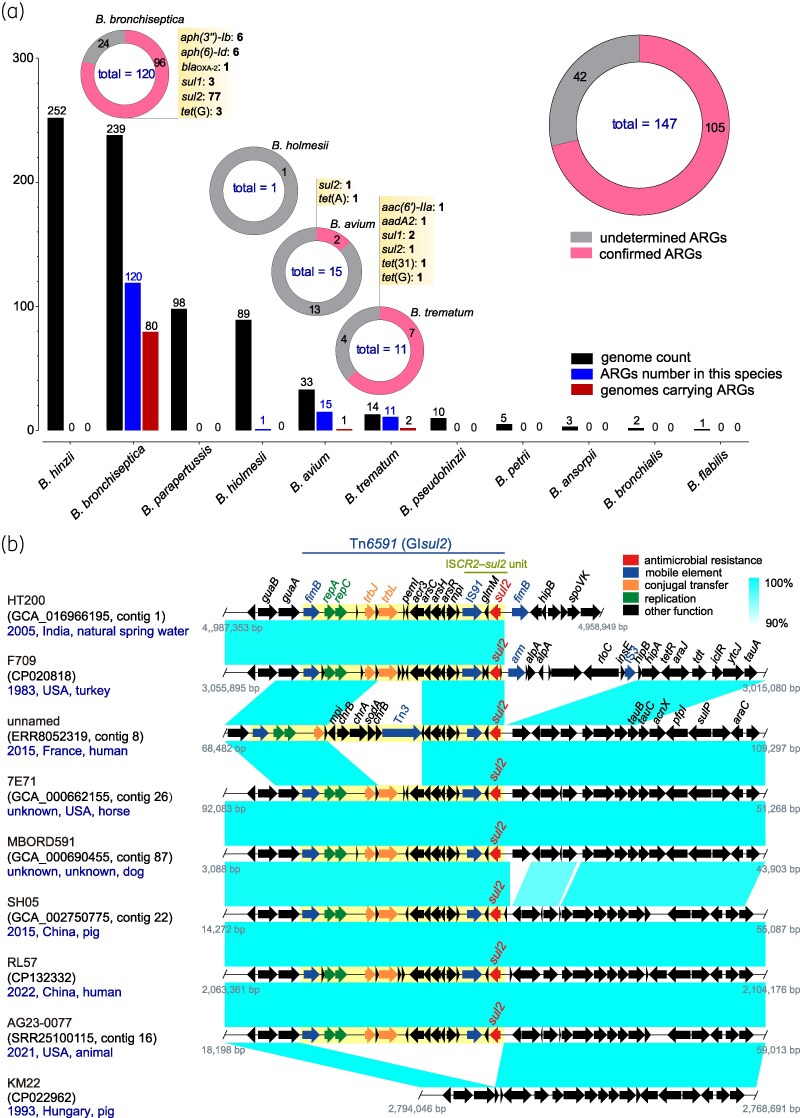 Figure 1
Figure 1 Figure 2
Figure 2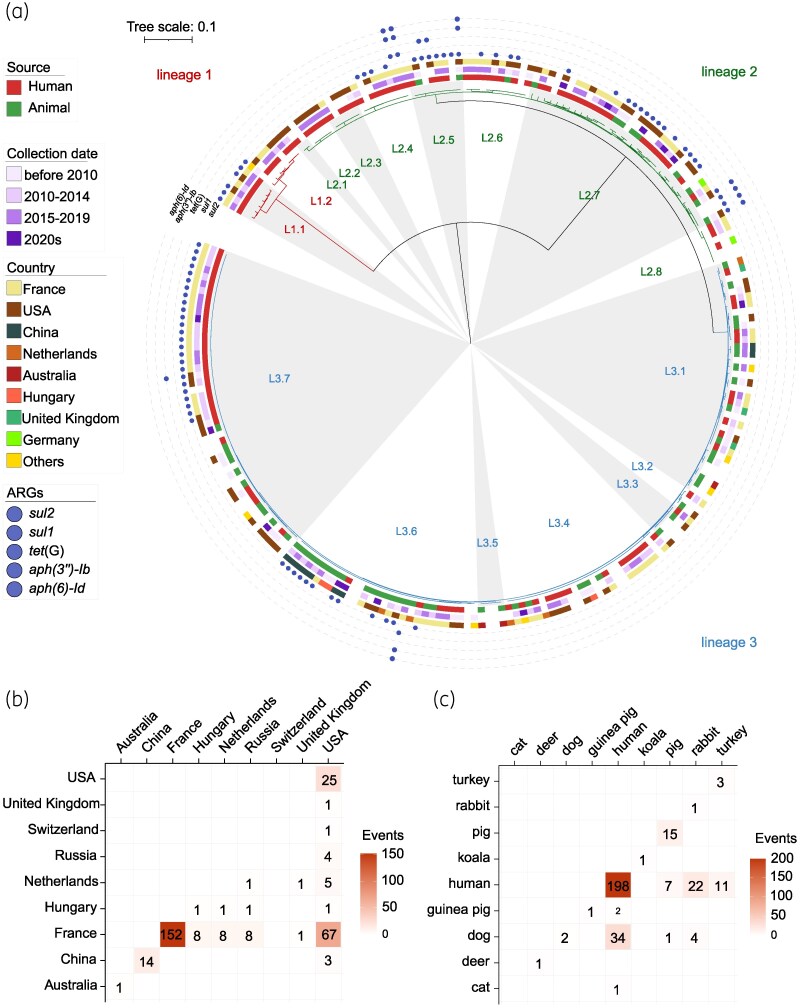 Figure 3
Figure 3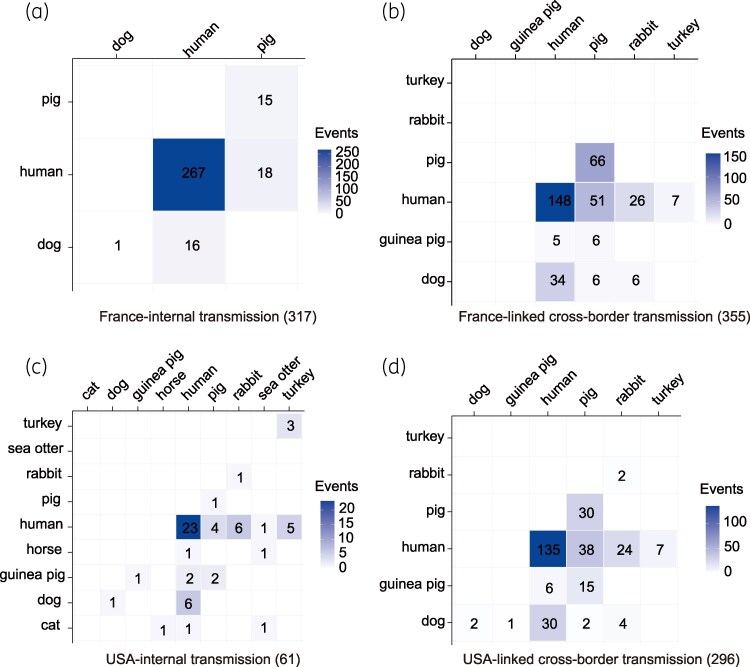 Figure 4
Figure 4- —‘Leading Goose’ R&D Program of Zhejiang Province10.13039/100022963
- —Zhejiang Provincial Natural Science Foundation of China10.13039/501100004731
- —Hangzhou Institute for Advanced Study
- —UCAS10.13039/501100015361
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Infections and Vaccines · Vector-borne infectious diseases · Diphtheria, Corynebacterium, and Tetanus
Introduction
Bordetella is a genus of Gram-negative bacteria, nearly all species of which can cause clinical infections in humans. The most commonly implicated species include B. pertussis, B. parapertussis, B. holmesii, and B. bronchiseptica.^1^ B. pertussis, the causative agent of pertussis (whooping cough), leads to severe acute respiratory infections. In recent years, other Bordetella species have also been increasingly reported in clinical cases.^2^ Given this trend, surveillance of antimicrobial resistance (AMR) in Bordetella spp. is crucial. Previous studies suggest that Bordetella spp. possess relatively conserved genomic sequences, which limit their ability to acquire antimicrobial resistance genes (ARGs) through horizontal gene transfer (HGT), particularly in B. pertussis.^3^ Our recent analysis corroborates these findings: among 8579 B. pertussis strains analysed, only 22 (0.26%) were predicted to carry ARGs, with the true frequency likely lower due to potential contaminant sequences.^4,5^ However, it remains unclear whether B. pertussis could acquire additional ARGs in the future.
HGT is more common among closely related bacterial species,^6^ raising concerns that other Bordetella species may serve as intermediaries, acquiring ARGs from external sources and potentially transmitting them to B. pertussis. However, the prevalence and genetic context of ARGs in Bordetella spp. (excluding B. pertussis) remain underexplored. Previous studies have reported the presence of acquired ARGs in B. bronchiseptica, for example in the works of Kadlec^7^ and Zhang^8^ et al., which were primarily based on PCR detection. These studies identified genes such as aac(6’)-Ib, sul, and tet.^7,8^ In contrast, reports of resistance genes in other Bordetella species are limited, leaving the broader distribution and evolutionary dynamics of ARGs within this genus largely unknown. To address this gap, we systematically investigated the distribution of ARGs in non-B. pertussis Bordetella species. This will provide critical insights into the potential risk of ARG acquisition in B. pertussis and inform strategies for managing AMR within the Bordetella genus.
Methods
Genome acquisition and quality control
Whole-genome sequences of Bordetella species analysed in this study were obtained from the NCBI database. We retrieved all available assembled genomes and high-throughput sequencing data as of October 28, 2024, selecting non-B. pertussis genomes (n = 1110). For 709 high-throughput sequencing datasets, we used Trimmomatic (v0.39; parameters: NexteraPE_TruSeq3-PE-2.fa:2:30:10:2:True SLIDINGWINDOW:4:15 LEADING:3 TRAILING:3 MINLEN:36) to remove adapters and low-quality sequences. The cleaned reads were assembled into genome contigs using SPAdes (v4.0.0; parameters: –isolate -k 21,33,55,77,99,127). We performed quality filtering of the 1110 genome sequences by ensuring the uniqueness of the corresponding BioSample. A total of 746 high-quality genomes were retained for downstream analyses based on four quality metrics: genome size, GC content, number of contigs, and N50 value. (Figure S1, available as Supplementary data at JAC Online).
Genome analysis
Of these, 329 were retrieved from NCBI assemblies, and 417 were obtained from SRA database (Table S1), which includes data from 11 Bordetella species (Figure 1a). The Table S1 also includes metadata from the database, with genomic source information validated and supplemented through relevant publications. To identify acquired ARGs, we used Abricate v1.0.1 (https://github.com/tseemann/abricate) and further analysed sequences using ResFinder 4.6.0 (http://genepi.food.dtu.dk/resfinder) and BLAST. Gene context for linear comparisons was visualized using Easyfig v2.2.3.
Analysis of acquired ARGs in the Bordetella genus and the genetic environment of the sul2 gene. (a) Prevalence of acquired ARGs in Bordetella species. (b) Genetic environment of the sul2 gene.
Phylogenetic analysis
The genomic sequences of the B. bronchiseptica were compared with the reference genomes NCTC10543 (GCA_900636925.1) using Snippy v4.3.6 (https://github.com/tseemann/snippy) with default parameters. A maximum-likelihood (ML) phylogenetic tree was inferred using IQ-TREE v1.6.12^9^ under the best-fit substitution model (selected by ModelFinder) with 1000 bootstrap replicates to assess branch support. Putative recombination regions were identified and removed using Gubbins v3.2.1.^10^ Population structure was further analysed through hierarchical Bayesian clustering implemented in RhierBAPS v1.0.1,^11^ which partitions strains into genetically distinct subgroups based on allelic variation. The phylogenetic tree was visualized and beautified using the online tool iTOL (https://itol.embl.de/).^12^
Inferring transmission events
To quantify the frequency of transmission events within and between different sources, we aggregated data over all B. bronchiseptica genomes and identified transmission events using a threshold-based approach based on SNP distances using a mapping procedure. SNPs were detected in the strain genome using snp-dists v.0.7.0 (https://github.com/tseemann/snp-dists).
Results and discussion
The prevalence of ARGs in non-B. pertussis Bordetella species
Among the 746 genome sequences, we identified 147 ARG copies across four Bordetella species: B. bronchiseptica (120), B. avium (15), B. trematum (11), and B. holmesii (1) (Figure 1a). However, due to the potential for exogenous sequence contamination, these numbers may not fully reflect the true ARG distribution. To solve this, we analysed the upstream and downstream sequences of the detected ARGs to exclude contamination, identifying 105 acquired ARGs likely to be genuinely present within the Bordetella genome. We confirm that three Bordetella species in this study harbour acquired ARGs: B. bronchiseptica, B. avium, and B. trematum. The prevalence of ARGs in these species was 33.6% (80/238), 3.0% (1/33), and 14.3% (2/14), respectively. Across these genomes, we identified 10 distinct ARG types spanning four antimicrobial classes. Sulphonamide resistance genes, particularly sul2, were the most prevalent, detected in all three species and found in 79 isolates, making it the dominant resistance genotype. Tetracycline resistance genes, such as tet(G), were also common, suggesting that the use of antibiotics and growth promoters in livestock and aquaculture may be key drivers of ARG acquisition in Bordetella. Aminoglycoside resistance genes exhibited the highest diversity, including aac(6’)-IIa, aadA2, aph(3'’)-Ib, and aph(6)-Id, while β-lactam resistance genes were rare, with blaOXA-2 detected only once.
It is important to emphasize that the in silico prediction of ARGs does not necessarily equate to their functional expression in Bordetella or the manifestation of a corresponding resistance phenotype. Multiple factors, including gene silencing, transcriptional regulation, and synergistic interactions, may modulate the phenotypic outcome. Therefore, comprehensive phenotypic validation of AMR is essential in future research.
The prevalence of the sul2 and tet(G) genes
The sul2 gene was most frequently observed in B. bronchiseptica. Analysis of its genetic environment suggests that its dissemination is likely driven by the mobile element Tn6591 (GIsul2) (Figure 1b), which serves as the core region. This element has been detected in B. bronchiseptica strains from multiple countries and diverse isolation sources, indicating its widespread presence.
For tet(G) in B. bronchiseptica, we identified two distinct genetic environments (Figure 2). In strain OSU-PE-IL99-1, tet(G) is embedded within a ∼44 kb gene fragment, with transfer likely mediated by a site-specific recombinase (Figure 2a). This fragment contains conjugative transfer-associated genes, and the presence of repA, a replication gene, suggests a plasmid origin. A tRNA-Arg sequence near the insertion site, known to be associated with genomic island (GI) formation and HGT,^13^ further supports this hypothesis. Additionally, highly homologous gene fragments containing the insertion site were detected in other Bordetella species, suggesting that tet(G) may be acquired through a similar mechanism in B. pertussis and other Bordetella species. In strain F1-PE-IL52-1, the tet(G) insertion site is located within tRNA-Gly and adjacent to tRNA-Cys (Figure 2b). Due to contig size limitations, part of the foreign fragment could not be definitively mapped to Bordetella sequences. Comparative analysis suggests that its transfer may be mediated by transposase, such as Tn3 or tniA, with the fragment likely originating from the Pseudomonas aeruginosa chromosome. Furthermore, homologous sequences containing the insertion site were detected in other Bordetella species, underscoring the broader risk of tet(G) acquisition.
Analysis of the genetic environment of the tet(G) gene in Bordetella. (a) Schematic of the integrative conjugative element harbouring tet(G) and other resistance genes inserted into the B. bronchiseptica genome. (b) The tet(G) gene and P. aeruginosa genomic sequences exhibit a shared genetic context.
The ARGs in B. bronchiseptica and B. trematum
In B. bronchiseptica, we identified ARGs such as sul1, blaOXA-2, aph(3'’)-Ib, and aph(6)-Id (Figure S2A). These genes co-occurred within a 58 kb nucleotide sequence detected in four genomes, one of which constitutes a complete plasmid, designated B. bronchiseptica plasmid R906, uploaded by Petrovski, S. in 2020. In addition to ARGs, this plasmid harbours genes conferring resistance to heavy metals, such as merA, merD, and merE. The plasmid belongs to the IncP1 replicon type, a broad-host-range plasmid widely distributed among Gram-negative bacteria.^14^ IncP1 plasmids have been shown to transfer and stably persist in B. pertussis under laboratory conditions.^15^ Moreover, a naturally occurring IncP1-type plasmid was reported in B. pertussis in 2002.^16^ Comparative analysis of plasmid-derived sequences from B. bronchiseptica revealed a highly similar plasmid backbone, suggesting that the Bordetella genus may rely on IncP1-type plasmids for acquiring resistance phenotypes.
Among the 14 B. trematum genomes analysed, two strains carried ARGs. One strain harboured tet(31) and sul2, while the other carried sul1, tet(G), floR, aadA1, and cmlA1 (Figure S2B). Comparative analysis with strain CBM01 revealed that the region containing ARGs in these two B. trematum genomes was integrated at the same chromosomal locus. In B. avium, HGT events were identified at two distinct chromosomal sites. Compared with the reference genome JBBA, strain BA8 acquired sul2 and tet(A) at separate loci (Figure S2C), suggesting independent HGT events.
Phylogenetic analysis based on core genome SNPs
To elucidate the dissemination of resistance determinants within the Bordetella genus, we performed a core-genome SNP-based phylogenetic analysis on 238 B. bronchiseptica isolates exhibiting the highest resistance-gene carriage. Bayesian clustering resolved the population into three principal lineages (lineage 1, lineage 2 and lineage 3; see Figure 3a), which were further subdivided into 17 sub-lineages. All isolates in lineage 1 originated from human hosts, whereas lineages 2 and 3 encompassed human, animal and environmental sources, indicating greater potential for cross-ecological transmission. No temporal clustering was observed among the three lineages, as isolates collected in different years exhibited comparable genetic distances, consistent with the known genomic conservation and low mutation rate of B. bronchiseptica. Among the 17 sub-lineages, strains from France and the United States were widely distributed, while Chinese isolates were predominantly assigned to sub-lineage L3.6, with a minority in L3.1. Analysis of resistance gene prevalence revealed that sul2, the most prevalent determinant, was unevenly distributed and particularly enriched in lineages L1.1, L2.4–L2.7, L3.6, and L3.7. Each of these sub-lineages includes human-derived isolates and thus represents a potential high-risk group. In addition, multidrug resistant isolates carrying multiple resistance genes were chiefly confined to lineage 2.
Phylogenetic Analysis and transmission characteristics of B. bronchiseptica. (a) Maximum likelihood phylogenetic tree of 238 B. bronchiseptica isolates. Scale bar represents nucleotide substitutions per site. (b) Heatmap depicting the number of inferred direct transmission events (SNP threshold < 5) between countries. Colour intensity is proportional to the number of events and does not account for the sample size per country. (c) Heatmap depicting the number of inferred direct transmission events (SNP threshold < 5) between host sources. Colour intensity is proportional to the number of events and does not account for the sample size per source.
Quantification of transmission events using SNP thresholds
To quantify and compare transmission dynamics, single SNP differences were classified as clonal transmission (SNPs < 5) or close/related transmission (5 ≤ SNPs < 20). Across all isolates, 492 clonal transmission events were identified, of which 304 had unambiguous information on country of isolation and host (Table S2). Among these 304 events, international transmission was frequent. Events involving France were the most numerous (152 domestic; 92 international). In contrast, for events involving the USA, international transmission constituted the majority (76.6%, 82/107) (Figure 3b). Numerous human-human events were detected, which may partly explain the high incidence of international spread. Cross-host transmission accounted for 27.0% (82/304) of events. Human-animal transmission comprised 77 cases, with human-dog (n = 34) and human-rabbit (n = 22) being the most common (Figure 3c). Expanding the threshold to SNPs < 20 uncovered additional transmission events, notably between the United States, China and France, and a marked increase in cross-host spread (Figure S3A). Human-pig close transmissions were most frequent (95 events) (Figure S3B).
Domestic versus international transmission patterns in France and the United States
Using SNPs < 20 as the criterion, domestic and international transmission events were tallied for the two principal source countries. France: 317 domestic versus 355 international events. Domestic spread was overwhelmingly human-human (84.2%, 267/317), with additional transmission observed between humans and dogs or pigs (Figure 4a). International events involved a broader host spectrum, though humans remained predominant (76.3%, 271/355); pigs, dogs and rabbits also featured prominently (Figure 4b). United States: 61 domestics (Figure 4c) versus 296 international events (Figure 4d), a pattern sharply contrasting with France. While domestic spread was predominantly human-human, the international profile resembled that of France, with human-centric transmission accompanied by significant pig, dog and rabbit involvement.
Transmission patterns centred on France and the USA with source composition of transmission events (SNPs < 20). (a) Local transmission events within France. (b) International transmission events linked to France. (c) Local transmission events within the USA. (d) International transmission events linked to the USA.
In both countries, international events outnumbered domestic ones—particularly in the United States. Dogs represent a principal host for B. bronchiseptica,^17^ and both France and the United States are major players in the global pet-dog trade.^18^ The international movement of dogs infected with specific clonal lineages could represent one of several possible contributors to international clonal dissemination. In the U.S. network, pet-trade-mediated human-dog/rabbit transmissions are frequently observed and may constitute a pathway through which international spread can exceed local transmission.
These findings indicate that human activity and the international movement of animals—particularly the pet-dog trade—are important routes for B. bronchiseptica dissemination, occasionally exerting greater influence than domestic transmission networks. This underscores the urgent need for stringent international pathogen control measures and enhanced international collaboration to prevent the spread of animal respiratory pathogens.
The number of Bordetella genomes analysed in this study is relatively limited and unevenly distributed across species. B. bronchiseptica is notably overrepresented, while genome sequences for other species remain scarce in public databases, likely contributing to the lower absolute number of ARGs detected in those taxa. These disparities may reflect both genuine differences in species prevalence and biases in genomic surveillance. To overcome these limitations, broader genome sequencing efforts across the genus are needed. We advocate for increased investment in this area, as integrating future datasets will be critical for more comprehensive assessments of AMR in Bordetella.
Conclusions
This work delivers the first comprehensive survey of acquired ARGs in non-B. pertussis Bordetella species, revealing ten ARG classes—including sulphonamide, tetracycline, aminoglycoside and β-lactam determinants—distributed across B. bronchiseptica, B. trematum and B. avium. Phylogenetic and transmission analyses demonstrate that, despite overall genomic conservation, B. bronchiseptica can integrate mobile elements carrying multiple ARGs, with evidence of clonal expansion and international, cross-host spread. The identification of plasmid- and transposon-mediated transfer underscores a risk for future ARG acquisitions in clinical and environmental Bordetella. These findings highlight the necessity for sustained, global genomic surveillance and collaboration to forestall the emergence and dissemination of resistant Bordetella strains.
Supplementary Material
dkaf418_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hu Y, Shi W, Meng Q et al Detection of Bordetella spp. in children with pertussis-like illness from 2018 to 2024 in China. J Infect 2024; 89: 106222. 10.1016/j.jinf.2024.10622239002934 · doi ↗ · pubmed ↗
- 2Mattoo S, Cherry JD. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin Microbiol Rev 2005; 18: 326–82. 10.1128/cmr.18.2.326-382.200515831828 PMC 1082800 · doi ↗ · pubmed ↗
- 3Linz B, Ivanov YV, Preston A et al Acquisition and loss of virulence-associated factors during genome evolution and speciation in three clades of Bordetella species. BMC genomics 2016; 17: 767. 10.1186/s 12864-016-3112-527716057 PMC 5045587 · doi ↗ · pubmed ↗
- 4Tang B, Hu X, Wu B et al Global antimicrobial resistance threats: insights from the resurgence of whooping cough. J Infect 2024; 89: 106337. 10.1016/j.jinf.2024.10633739481609 · doi ↗ · pubmed ↗
- 5Zhuang Y, Li S, Bao D et al Genomic integrity in Bordetella pertussis: avoiding contaminant-derived misinterpretations of acquired antimicrobial resistance. J Infect 2024; 89: 106356. 10.1016/j.jinf.2024.10635639557090 · doi ↗ · pubmed ↗
- 6Arnold BJ, Huang IT, Hanage WP. Horizontal gene transfer and adaptive evolution in bacteria. Nat Rev Microbiol 2022; 20: 206–18. 10.1038/s 41579-021-00650-434773098 · doi ↗ · pubmed ↗
- 7Kadlec K, Schwarz S. Antimicrobial resistance in Bordetella bronchiseptica. Microbiol Spectr 2018; 6: 10.1128/microbiolspec.arba-0024-2017. 10.1128/microbiolspec.ARBA-0024-2017 · doi ↗
- 8Zhang Y, Yang H, Guo L et al Isolation, antimicrobial resistance phenotypes, and virulence genes of Bordetella bronchiseptica from pigs in China, 2018–2020. Front Vet Sci 2021; 8: 672716. 10.3389/fvets.2021.67271634169108 PMC 8217433 · doi ↗ · pubmed ↗