Case Report: Association of a rare single nucleotide variant in the KCNH2 gene with drug-induced QT prolongation
Tianci Wang, Charlene R. Norgan Radler, Mohanakrishnan Sathyamoorthy

TL;DR
A rare genetic variant in the KCNH2 gene is linked to drug-induced QT prolongation, a heart condition that can lead to dangerous arrhythmias.
Contribution
This is the first study to associate the KCNH2 c.1066C>T variant directly with drug-induced QT prolongation and assess its pathogenicity using ACMG guidelines.
Findings
The KCNH2 c.1066C>T variant was found in a patient with drug-induced QT prolongation.
In silico analysis suggested potential pathogenicity, but structural impacts remain uncertain.
The variant was classified as 'uncertain significance' based on ACMG and ClinGen criteria.
Abstract
Long QT Syndrome (LQTS) is characterized by prolonged QT intervals on electrocardiogram, which may progress into life-threatening polymorphic ventricular tachycardia and sudden cardiac death. Variants in the KCNH2 gene have been associated with congenital LQTS, with thousands identified to date but very few clinically characterized. To describe the rare single nucleotide variant KCNH2 (NM_000238.4):c.1066C>T (p.Arg356Cys) associated with drug-induced QT prolongation and to assess its pathogenicity risk using in silico tools and protein structural modeling in accordance with American College of Medical Genetics and Genomics (ACMG) guidelines. Next-generation sequencing was performed for a patient presenting with drug-induced QT prolongation who was found to carry the rare KCNH2 1066C>T variant. Thirteen established gene discovery computational tools were employed to analyze the variant…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
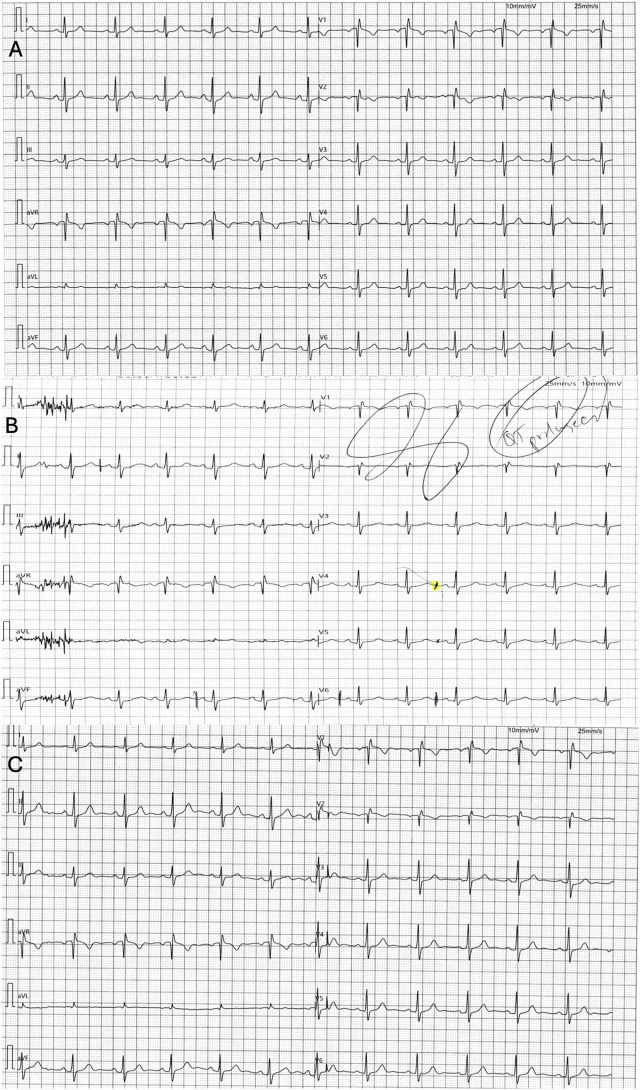 FIGURE 1
FIGURE 1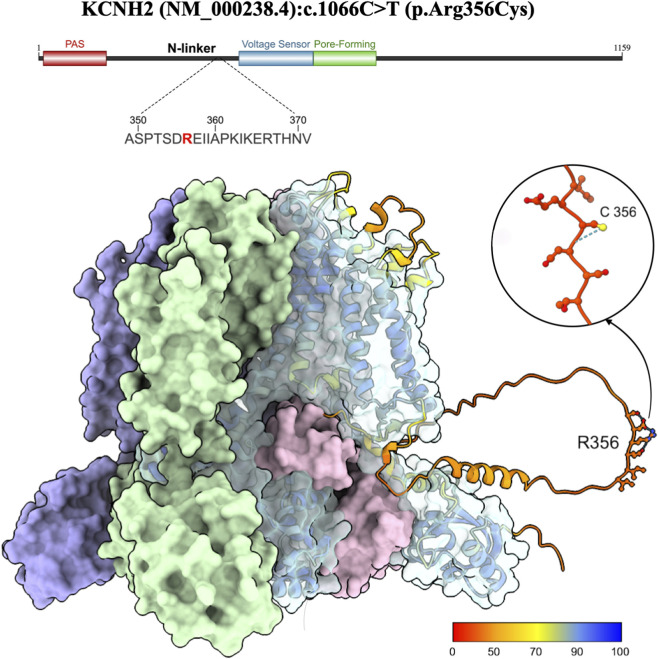 FIGURE 2
FIGURE 2| Evidence type | Benign strong | Benign supporting | Pathogenic supporting | Pathogenic moderate | Pathogenic strong | Pathogenic very strong |
|---|---|---|---|---|---|---|
| Population Data | gnomAD FAF (0.004382%) does not support | | | gnomAD FAF (0.004382%) does not support | Observed in an unrelated 8 y.o. female with LQTS (QTc 450 ms) and a | |
| Computational and Predictive Data ( | | REVEL score (0.659) does not support | REVEL score (0.659) is PP3 supporting | A different pathogenic variant does not exist at the same residue. Does not support | An established pathogenic variant with same amino acid change does not exist. Does not support |
|
| Functional Data ( | One RNA/protein metabolism assay showed no damaging effect. ( | | Missense constraint Z-score is 2.5, thus benign missense variation is not significantly depleted. Does not support | Not located in a mutational hotspot. Does not support | Functional studies have not shown a damaging effect. Does not support | |
| Segregation Data | Biological family member data is unavailable. In one unrelated family, the proband’s reportedly unaffected father had a VUS in both | |||||
|
| | | | Data is not available from the biological family and does not support | | |
| Allelic Data | | Not observed in | |
| | |
| Other | | An alternate molecular basis for disease was not found among 42 genes tested. Does not support | Schwartz-score ( | | | |
| VARIANT CLASSIFICATION: UNCERTAIN SIGNIFICANCE (1 point) | ||||||
| Tool | Benign very strong | Benign strong | Benign moderate | Indeter-minate | Pathogenic supporting | Pathogenic moderate | Pathogenic strong | Pathogenic very strong |
|---|---|---|---|---|---|---|---|---|
| Supervised machine learning | ||||||||
| BayesDel | | | 0.057 | | | | | |
| CADD v1.7 | | | | | 27.6 | | | |
| REVEL | | | | | 0.659 | | | |
| VEST4 | | | 0.601 | | | | ||
| Structural/Physicochemical parameters | ||||||||
| Evolutionary Action | | | 0.467 | | | | ||
| MutPred2 | | | 0.341 | | | | | |
| PolyPhen2 | | | | 0.998 | | | ||
| Sequence conservation | ||||||||
| PhyloP | | | 3.357 | | | | ||
| PrimateAI | | 0.534 | | | ||||
| FATHMM | | −2.33 | | | ||||
| GERP++ | | | | |||||
| SIFT | | 0.04 | | | | |||
| Organism | Protein ID | Alignment | Similarity |
|---|---|---|---|
|
| 346 D P F L A S P T S D | 100% | |
|
| 388 D P F L A S P T S D | 100% | |
|
| 287 D P F L A S P T S D | 100% | |
|
| 344 D P F L A S P T S D | 100% | |
|
| 346 D P F L A S P T S D | 100% | |
|
| 348 D P F L A S P T S D | 100% | |
|
| 331 D P F L A S P T S D | 100% | |
|
| 359 D P F L A S P T S D | 100% | |
|
| 354 D P F L A S P T S D | 100% | |
|
| 354 D P F L A S P S S D | 97% | |
|
| 343 D P F L A S P T S D | 100% | |
|
| 259 D T F L A T P S G E | 44% | |
|
| 344 D A F L G A P S G E | 41% | |
|
| 286 D A F L A A P P G E | 42% |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac electrophysiology and arrhythmias · ECG Monitoring and Analysis · Cardiac Arrhythmias and Treatments
Introduction
Long QT syndrome (LQTS) is characterized by delayed cardiac repolarization seen as a prolonged QT interval on electrocardiogram (ECG). After heart rate correction, a prolonged QT interval (QTc) is considered as >460 ms in females, and >450 ms in males (Krahn et al., 2022). LQTS is dangerous due to its potential to progress into torsades de pointes, a life-threatening polymorphic ventricular tachycardia. LQTS is diagnosed by either an isolated QTc ≥500 ms, or Schwartz score ≥3.5 points, which incorporates the degree of QT prolongation, other ECG abnormalities, clinical history, and family history (Krahn et al., 2022; Schwartz and Ackerman, 2013). The most common presentation of LQTS is recurrent syncope, although patients may range from asymptomatic to experiencing dizziness, palpitations, dyspnea, or sudden cardiac death (Krahn et al., 2022).
LQTS may be congenital or acquired. Congenital LQTS consists of heritable diseases caused by abnormal cardiac ion channel function, affecting about 1 in 2,000-2,500 live births (Schwartz et al., 2009). Over 15 subtypes of congenital LQTS have been identified, based on the affected ion channel gene (Wilde et al., 2022). Subtypes 1–3 account for over 75% of cases and are caused by variants in KCNQ1, KCNH2, and SCN5A, respectively. Other, less commonly genes include KCNE1, KCNE2, CACNA1C, CALM1-3, and CAV3 (Wilde et al., 2022). Congenital LQTS is known for its clinical heterogeneity and variable penetrance, complicating timely diagnosis and management (Krahn et al., 2022).
Acquired LQTS is more common and results from factors such as electrolyte disturbances, medications, or bradyarrhythmias (El-Sherif et al., 2018). The most frequent mechanism involves drug-induced blockade of the voltage-gated inwardly rectifying potassium channel encoded by KCNH2 (El-Sherif et al., 2018). Despite preclinical screening for inhibition of this potassium current during drug development, many commonly used medications, such as antipsychotics, antibiotics, and antiarrhythmics, still carry QT-prolonging risk (Roden, 2016). Importantly, about a third of acquired LQTS patients have been found with pathogenic variants in LQTS-causing genes (Itoh et al., 2016). This suggests that medications may unmask an underlying genetic predisposition to LQTS, rather than being the sole cause of QT prolongation (Itoh et al., 2016).
The voltage-gated inwardly rectifying potassium channel KCNH2 conducts the rapid delayed rectifier potassium current (I Kr) during cardiac repolarization. It is a tetramer composed of a cytosolic N-terminus containing a PAS (Per-Arnt-Sim) domain, an N-linker, six transmembrane helices containing the voltage sensor, a selectivity filter, channel pore, and a cytosolic C-terminus containing a cyclic nucleotide-binding homology domain (CNBHD). The CNBHD does not bind any ligands but physically interacts with the PAS domain intracellularly to regulate channel gating (Foo et al., 2016). In native cardiac tissue, functional channels are hetero-tetramer of 1a and 1b subunits. Subunit 1a is the full-length channel protein, whereas 1b is an alternatively-spliced isoform replacing most cytosolic N-terminal domains with a unique 35-amino acid sequence. Co-assembly of both subunits is essential for channel function and cardiac repolarization (Foo et al., 2016).
KCNH2 remains a major focus of LQTS research due to its implications in both congenital and acquired disease, with thousands of variants identified to date (Liu et al., 2025). In this study, we describe a patient without prior cardiac history who developed drug-induced LQTS and was found to carry a rare KCNH2 variant of uncertain significance (VUS). In silico analyses and computational modeling of the region of interest were performed as preliminary assessment of the variant’s impact. A risk assessment was conducted based on American College of Medical Genetics and Genomics (ACMG) guidelines and ClinGen recommendations.
Materials and methods
Genetic testing
We utilized a commercially available panel targeting 42 genes associated with inherited arrythmias including LQTS, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, arrhythmogenic right ventricular cardiomyopathy, along with other arrhythmias/channelopathies and sudden cardiac arrest (Supplementary Table S1) (Ambry Genetics, 2025). Details of the sequencing methods have been published previously and are provided in the Supplementary Material (Ambry Genetics, 2025).
Variant analysis and interpretation
Information regarding the structure of voltage-gated inwardly rectifying potassium channel KCNH2 was obtained from UniProt (Nucleic Acids Research, 2025). Population frequency and ancestry-specific data were retrieved from Genome Aggregation Database (GnomAD), an international database aggregating data from a wide variety of large-scale data sequencing projects (Karczewski et al., 2020). All KCNH2 N-linker variants were evaluated using public databases, including Human Gene Mutation Database (HGMD), Online Mendelian Inheritance in Man (OMIM), and ClinVar to assess phenotypic associations, pathogenicity evidence, and evolutionary conservation (National Library of Medicine, 2025; Stenson et al., 2014; Johns Hopkins University, 2025).
The Grantham score was used to predict the potential impact of the single amino acid substitution of the variant (Grantham, 1974). Additionally, variant risk interpretation was performed using American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines and Clinical Genome Resource (ClinGen) criteria-specific recommendations. Variant pathogenicity was classified using REVEL, and additional analyses performed using calibrated in silico tools (Tavtigian et al., 2008; Ioannidis et al., 2016; Tavtigian et al., 2006; Mathe et al., 2006; Ng and Henikoff, 2001; Choi et al., 2012; Choi and Chan, 2015; Shihab et al., 2013; Davydov et al., 2010; Cooper et al., 2005; Pollard et al., 2010; Sundaram et al., 2018; Katsonis and Lichtarge, 2014; Adzhubei et al., 2010; Kircher et al., 2014; Schubach et al., 2024; Feng, 2017; Carter et al., 2013; Pejaver et al., 2022; Pejaver et al., 2020), for which methodology has previously been described by our group (Norgan Radler et al., 2025). The selected tools along with their classification thresholds, mechanistic principles, clinical recognition, and limitations are provided in Supplementary Table S2.
The cryogenic electron microscopy (cryo-EM) assembly structure for the voltage-gated inwardly rectifying potassium channel KCNH2 (5VA2) was obtained from Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) (Wang and MacKinnon, 2017; Berman et al., 2003). The monomeric wild-type predicted protein structure (Q12809) was obtained from AlphaFold Protein Structure Database (AFDB) and overlaid on a single monomer of the cryo-EM structure 5VA2 using the UCSF ChimeraX matchmaker tool (Varadi et al., 2022; Varadi et al., 2024). This method enables visualization of the arginine at position 356, which is absent from experimental structures in RCSB PDB.
The structure of KCNH2 encoded protein containing the variant was predicted in AlphaFold3 using the NCBI FASTA reference sequence NM_000238.4, for which detailed methodology has been described by our group. Detailed data of AlphaFold modeling including confidence measures for the overall structure and local residues are detailed in Supplementary Table S3.
Informed consent was obtained from the individual participant involved in the study and is on file at CCMS-FW. Institutional Review Board approval was waived for this study.
Results
Clinical features
A 48-year-old female with a medical history of Crohn’s disease status post ileostomy was initially referred to our clinic for evaluation of a first degree atrial-ventricular block and premature ventricular contractions (PVC) noted on a pre-operative electrocardiogram (ECG) for ileostomy. She reported occasional palpitations but denied other cardiac symptoms or cardiac history including arrhythmias, coronary artery disease, or congestive heart failure. The patient was adopted with no available family medical history.
Initial in-office ECG showed right bundle branch block (RBBB) with a qR pattern in lead V1, normal axis, and a normal QTc (Figure 1, top panel). Transthoracic echocardiography demonstrated a normal left ventricular ejection fraction (>70%) without evidence of right ventricular hypertrophy or indirect features of pulmonary arterial hypertension. A 48-h Holter monitor revealed a premature ventricular contraction and premature atrial contraction (PAC) burden of less than 1%, without any sustained atrial or ventricular arrhythmias.
Patient ECGs at baseline (top) and during the long QT episode (middle) and after discontinuation of Ciprofloxacin (bottom). (A) Initial/baseline ECG obtained 3 months before the episode demonstrated sinus rhythm with early precordial qR transitions with a slightly elevated ST segment with a normal corrected QT interval of 435 ms. (B) 12-lead resting baseline ECG for stress test (note lead representation configured for stress ECG) demonstrated prolonged QT interval (QTc 545 ms). The patient had started ciprofloxacin 500 mg three times daily 5 days prior to this tracing. (C) ECG obtained 5 days after discontinuation of ciprofloxacin, demonstrating normal sinus rhythm with normalization of the QT interval to baseline (QTc of 428 ms).
The patient returned a few months later for further evaluation of a chest pounding sensation with dizziness and palpitations. She underwent a stress echocardiogram for age-appropriate ischemia risk stratification. Her stress ECG was negative for ischemia, but the corrected QT interval was noted to be significantly prolonged to 545 ms by Bazett formula (Figure 1, middle panel), which was not evident on her baseline ECG (Figure 1, top panel). We learned that she had recently initiated ciprofloxacin (5 days prior) and metronidazole for treatment of small intestine bacterial overgrowth. No further changes in medications were identified, and her Crohn’s disease was managed with as-needed hyoscyamine following her ileostomy. A comprehensive metabolic panel (CMP) ordered immediately showed no electrolyte abnormalities, with a normal potassium level of 4.1 mmol/L, and a calcium level of 9.4 mmol/L. The patient was known to have a normal magnesium level through review of historical labs. She was advised to immediately discontinue ciprofloxacin and educated on QT-prolonging agents. Magnesium oxide was initiated for stabilization of myocardial repolarization, and a mobile cardiac outpatient telemetry (MCOT) device was used for continuous ambulatory monitoring.
Following discontinuation of ciprofloxacin, ventricular arrhythmias or QT prolongation were not recorded during a 7-day MCOT monitoring period. In clinical follow-up at 1 week, her repeat ECG demonstrated normalization of the QT interval to her original baseline (Figure 1, bottom panel). Given the episode of a definitive drug-induced QT prolongation, we advised hypervigilant surveillance for any QT-prolonging medications and proceeded with clinical genetic testing to evaluate for any high risk pathogenic channelopathies. Biological family members were not available to provide additional clinical information or undergo family genetic studies.
Genetic analysis
Genetic sequencing revealed a single variant of uncertain significance (VUS) in KCNH2 (NM_000238.4):c.1066C>T (p.Arg356Cys). KCNH2 has been definitively associated with long and short QT syndromes by Clinical Genome Resource (ClinGen) Gene Curation Expert Panels (GCEPs) (Clinical Domain Working Groups, 2025; Harrison et al., 2019; Richards et al., 2015; Clinicalgenome, 2025). A pathogenicity risk analysis was performed utilizing American College of Medical Genetics and Genomics (ACMG) guidelines along with ClinGen criteria-specific recommendations, with a resulting overall classification of “uncertain significance” (Table 1). The evidence utilized for each ACMG criterion is listed below.
Population data
The gnomAD GroupMax filtering allele frequency (FAF) for KCNH2 (NM_000238.4):c.1066C>T is 0.004382% among South Asians, suggesting it is a very rare variant. The FAF is the maximum credible genetic ancestry group allele frequency (Harrison et al., 2019). Public databases were investigated for reports of the variant in unrelated patients with LQTS since case-control studies may not reach statistical significance for very rare variants. ClinVar contains five submissions for the variant, four of which are associated with LQTS or cardiac arrhythmias with unknown affected status (Supplementary Table S4) (National Library of Medicine, 2025). While the variant has also been reported in an unrelated eight-year-old female with LQTS (QTc 450 ms), she was found to have an additional KCNQ1 VUS, resulting in uncertainty regarding each variant’s contribution. Although both variants were also carried by the patient’s reportedly unaffected father, we assessed this observation is not benign-supporting since LQTS has incomplete penetrance.
Computational and predictive data
Computational data supports variant pathogenicity using a REVEL score of 0.659 per ClinGen recommendations for PP3 criteria (Supplementary Table S2). Additional in silico analyses were performed using well-established gene discovery tools to investigate conservation and predicted structural and functional impacts (Table 2) (Garcia et al., 2022).
Conservation analysis demonstrated that the arginine residue and its surrounding region are highly conserved (97%–100% identical) across nearly all examined mammalian species (Table 3). In non-mammalian species included in our analysis (birds and reptiles), this region showed significant divergence, although the arginine residue is consistently replaced by lysine, an amino acid with similar biochemical properties. Notably, the R residue is flanked by two acidic residues, glutamic acid and aspartic acid in mammals, and two glutamic acids in non-mammalian species, maintaining the local electrostatic environment. However, all four in silico tools that assess evolutionary sequence conservation (PrimateAI, PhyloP, SIFT, FATHMM) classified this variant as “indeterminant.”
The amino acid substitution of arginine to cysteine has a Grantham Score of 180 and is classified as a “radical change.” However, this variant was predicted as “likely benign” with a score of 0.312 by AlphaMissense, a deep-learning-based sequence analysis integrating structural context and population frequency. Other tools such as MutPred2 and PolyPhen-2 produced conflicting analyses of structural and functional consequences in the protein. Intriguingly, AlphaFold3 modeling revealed the variant is in a disordered region that is not well-characterized experimentally (Figure 2). The discordance between the amino acid substitutions and their predicted structural impact may partially result from the uncertain conformation of disordered regions. Detailed data of AlphaFold modeling are provided in Supplementary Table S3. Of note, the predicted structures include disordered regions in the N-linker, extracellular loops, and C-terminus which are usually excluded from experimental models and lower the global pLDDT.
Structural visualization of the voltage-gated inwardly rectifying potassium channel KCNH2 from the RCSB PDB biological assembly 5VA2 (Wang and MacKinnon, 2017; Xie et al., 2022). The corresponding full-length predicted AFDB structure Q12809 overlays a single monomer, colored by per-atom predicted Local Distance Difference Test (pLDDT). Arginine at position 356 is labeled and visualized using ball stick style with heteroatom coloring. A color key located at the bottom right of the figure depicts pLDDT values, with higher values demonstrating higher confidence in the predicted structure.
Functional data
The surrounding N-terminal linker region, which is not a known mutational hotspot, has 344 variants reported in ClinVar, most frequently in association with LQTS, followed by cardiovascular phenotype, cardiac arrythmia, and short QT syndrome (Supplementary Table S5). Most of the variants associated with LQTS have an unknown affected status and are classified as either VUS or as having conflicting classifications. Only three variants in this region are classified as likely pathogenic for LQTS; KCNH2 916G>T and 916G>C, which localize in a consensus splice site, and KCNH2 1015A>T. The variants KCNH2 916G>T and 916G>C have both been reported in patients with clinically confirmed LQTS in large-cohort sequencing studies, while KCNH2 1015A>T has been observed in a single patient with LQTS (Giudicessi et al., 2012; Tester et al., 2005). Human Gene Mutation Database (HGMD) reported 70 variants in the N-linker region, with similar associated phenotypes of LQTS and short QT syndrome, and additional conditions including sudden cardiac death, Brugada syndrome, sudden cardiac death, bicuspid aortic valve, sudden infant death, and catecholaminergic polymorphic ventricular tachycardia. Most variants were classified as disease-causing mutations (DM) for LQTS and Brugada syndrome (Supplementary Table S5) (Qiagendigitalinsights, 2025).
Segregation/de novo/allelic data
LQTS is inherited in an autosomal dominant pattern with incomplete penetrance. The variant was not assessed for segregation with LQTS or de novo status because the patient’s biological family was unavailable. In one unrelated family, the proband’s reportedly unaffected father carried a VUS in both KCNH2 and KCNQ1 although cardiovascular diagnostic findings were not available (Bora et al., 2023). The variant was not observed in cis or trans with another variant.
Other data
A Schwartz-score of zero was calculated based on: QTc 545 ms after exercise stress test in presence of QT-prolonging agent (Krahn et al., 2022; Priori et al., 2013). The patient did not have a history of syncope or congenital deafness, and has unknown biological family history. An alternate molecular basis for disease was not found among 42 genes tested.
Discussion
In this study, we present novel clinical data on a rare single nucleotide variant, KCNH2 (NM_000238.4):c.1066C>T, associated with drug-induced QT prolongation in a patient with no prior cardiac history, with in silico analyses and computational modeling to visualize the variant location and potential impact. With recent development of high-throughput automated expression and functional assessment assays for KCNH2 variants, reporting of granular, variant-level clinical data remains valuable to assist in clinical risk stratification and validation of in vitro findings.
Loss-of-function variants in KCNH2 may impair protein synthesis, trafficking, channel gating, or ion permeability, with over 80% resulting in trafficking defects (Delisle et al., 2004; Anderson et al., 2006; Anderson et al., 2014). Interestingly, variant impact appears to be influenced by domain location. Variants affecting the channel pore, particularly the S5-S6 helices, typically cause severe trafficking defects and often exert dominant-negative suppression of the WT protein, an effect less frequently observed in other domains. In contrast, N-terminal PAS domain and S4-S5 linker variants tend to alter channel gating, whereas C linker/CNBHD domain variants exhibit a mix of trafficking or gating defects depending on the specific locus affected (Foo et al., 2016; Anderson et al., 2006; Anderson et al., 2014). Clinically, pore-region variants are associated with a higher incidence of arrhythmic events than non-pore regions (Shimizu et al., 2009; Moss et al., 2002). A retrospective analysis of 2,826 KCNH2 variants from publicly available gene databases showed that variants localized in the S5-S6 region including the selectivity filter, as well as the PAS domain, are associated with increased risk of cardiac events including torsades de pointes and sudden cardiac death (Liu et al., 2025).
This variant lies within the N-linker region which lacks a highly ordered structure and is generally considered more tolerant of amino acid changes (Anderson et al., 2014). The disordered structure also creates challenges in predicting local interactions and structural alterations via computational modeling. However, REVEL, a meta-predictor validated for clinical variant classification, provided evidence supporting pathogenicity based on ClinGen calibrated thresholds. Collectively, the combined clinical, population and computational evidence in this study supports a classification of “uncertain significance” for the variant.
To date, one prior report identified this variant in an eight-year-old female with a QTc of 450 ms who presented with dyspnea and palpitations (Bora et al., 2023). Notably, the patient also carried a VUS in KCNQ1 (associated with LQT1) and both variants were also identified in the patient’s reportedly unaffected father (Bora et al., 2023). Additionally, a different variant affecting the same amino acid residue, KCNH2 1067G>A, was reported along with another variant, KCNH2 1682C>T, in a family with LQTS and sudden cardiac death, with both variants strongly segregating with the phenotype (Riuró et al., 2015). A recent high-throughput cell-surface expression study examined over 18,000 KCNH2 variants including our variant of interest, and did not identify significant changes in cell surface expression compared with the wild-type (O’Neill et al., 2024). To our knowledge, variant-specific data is not available regarding effects on current conduction, gating kinetics, or interaction dynamics with wild-type 1a and 1b subunits, all of which are key parameters for understanding the variant’s functional consequences.
The N-terminal region, including the PAS domain and N-linker, has been implicated in regulating channel deactivation by physically interacting with the CNBHD domain, and facilitating the co-assembly of 1a and 1b subunits (Foo et al., 2016; Liu et al., 2016; Phartiyal et al., 2007). In vitro mechanistic studies on the N-linker region have been limited to segments that do not include the region harboring our variant, reducing their direct relevance to our work; nevertheless, several N-linker variants have been functionally characterized (Johnson et al., 2022). A 2009 study functionally examined eight variants identified in drug-induced LQTS patients, including an N-linker variant, KCNH2 1025A>T, found in a 70-year-old woman with markedly prolonged QTc (776 ms) and torsades de pointes following erythromycin exposure (Itoh et al., 2009). In Chinese Hamster Ovarian (CHO) cells, KCNH2 1025A>T did not have a detectable current when expressed alone and exerted a mild dominant-negative effect on the WT subunit when co-expressed (Itoh et al., 2009). Abnormal gating was also observed including a negative shift in the voltage dependence of inactivation and accelerated inactivation kinetics (Itoh et al., 2009). Similar patterns of mildly reduced current density and gating defects have been reported for other N-linker variants, partially consistent with a recent large-scale study suggesting variants in this region generally produce mild trafficking and conduction defects (O’Neill et al., 2024; Mattivi et al., 2020; Hayashi et al., 2020; Fodstad et al., 2006).
Heritable arrhythmogenic disorders affecting cardiac ion channels, such as LQTS and Brugada Syndrome, are generally transmitted in an autosomal dominant pattern with exceptions including Jervell and Lange-Nielsen syndrome. These cardiac channelopathies may demonstrate incomplete penetrance in which some individuals with a disease-causing variant do not manifest associated phenotypic features. Prior population-based estimates of LQTS penetrance are near 40%, with even highly penetrant founder mutations demonstrating phenotypic variability (Berge et al., 2008; Brink et al., 2005). Potential non-genetic modifiers contributing to these observed differences include sex and age, with males demonstrating greater risk of sudden cardiac death (Coll et al., 2017; Giudicessi and Ackerman, 2013). Age-related penetrance was previously demonstrated among children undergoing ajmaline provocation for Brugada Syndrome (McMillan et al., 2014; Conte et al., 2014). Additionally, exogenous factors such as excessive alcohol intake may unmask phenotypic electrocardiogram patterns in Brugada Syndrome. The incomplete penetrance and age-dependent expression observed in autosomal dominant cardiac channelopathies lend complexity to interpreting VUS findings in cases that may initially appear clinically silent.
In this case, the patient reported palpitations and dizziness which are a common presentation of LQTS, although many patients may remain asymptomatic (Millat et al., 2006). Given the risk of sudden cardiac death associated with LQTS and the widespread use of QT-prolonging medications, clinicians should maintain a high index of suspicion and pursue genetic screening in suspected cases or for first-degree relatives of affected cases (Al-Khatib et al., 2017). In accordance with the 2017 AHA/ACC/HRS guideline for the management of patients with ventricular arrythmias, individuals carrying the KCNH2 1066C>T variant should avoid QT-prolonging medications when possible; if required, careful monitoring of the QTc during treatment is needed with prompt discontinuation if marked QTc prolongation develops (Al-Khatib et al., 2017). Beta-blocker therapy is strongly recommended for patients with a resting QTc >470 ms, and is considered reasonable as long-term therapy in asymptomatic patients with resting QTc <470 ms (Al-Khatib et al., 2017). In patients with a significant symptom burden or symptoms refractory to beta-blocker treatment, more invasive therapy such as sympathectomy and implantable cardioverter defibrillator should be considered (Al-Khatib et al., 2017; Hauwanga et al., 2025).
Long-term clinical follow up and education on avoidance of QT-prolonging agents are important for all patients with LQTS, and individuals carrying variants of uncertain significance associated with LQTS, particularly when preliminary analyses such as in silico predictions suggest potential pathogenicity (Richards et al., 2015). The importance of continued surveillance is underscored by the population-level observation that the risk of sudden cardiac death increases steadily with age until reaching 60–80 years (Stecker et al., 2014; Stiles et al., 2020), likely reflecting the combined effects of underlying channelopathies and the increasing prevalence of cardiac comorbidities such as ischemic heart disease. Long-term clinical follow-up allows patients to receive updated information regarding potential variant reclassification (Muller et al., 2020), and also enables accurate and ongoing risk assessment as additional family and medical history emerge.
The QT prolongation in our patient was most likely triggered by ciprofloxacin, given its known QT-prolonging risks, and the observed strong temporal association and prompt resolution following its discontinuation. The patient’s other medications included metronidazole and hyoscyamine. While metronidazole carries a conditional QT-prolonging risk in the setting of excessive dosing, electrolyte abnormalities, or drug-drug interactions, these factors were not identified in this case (Wu et al., 2025; Crediblemeds, 2025). The potential contribution of several additional factors cannot be fully excluded. Notably, a serum magnesium level was not obtained at the time of her presentation. Isolated hypomagnesemia is a recognized cause of QT-prolongation in both healthy adults and susceptible populations (Moulin et al., 2015; Kieboom et al., 2016). Furthermore, mental stress, sympathetic activation, and autonomic conflict have also been shown to induce transient QT-prolongation, T wave abnormalities and arrythmias, particularly in genetically susceptible patients. Such factors could not be assessed in this case (Noda et al., 2002; Winter et al., 2018; Andrássy et al., 2007).
We acknowledge several limitations. Firstly, family pedigree and co-segregation analyses could not be performed due to the patient’s adopted status. Secondly, the reported genotype-to-phenotype association is based on a single patient and is limited by the potential influence of factors described above as well as other unidentified genetic modifiers. Thirdly, although in silico analyses offer valuable insight, critical information from electrophysiological data is not available. Future studies should explore in vitro expression and functional assessment of the variant via cell-surface ELISA, Western blotting and whole-cell patch-clamp to evaluate its impact on channel synthesis, trafficking, and function, to help clarify the mechanistic link between the genetic variant and the observed clinical phenotype.
Conclusion
To our knowledge, this is the first study with detailed clinical data linking the rare variant KCNH2 1066C>T to drug-induced QT prolongation. Initial risk assessment of this variant utilizing population data and in silico analyses resulted in an overall classification of “uncertain significance” based on ACMG and ClinGen guidelines. Further functional characterization studies are warranted to further validate the pathogenicity of this variant and elucidate the mechanism of its phenotypes observed in this patient.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adzhubei I. A. Schmidt S. Peshkin L. Ramensky V. E. Gerasimova A. Bork P. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 (4), 248–249. 10.1038/nmeth 0410-248 20354512 PMC 2855889 · doi ↗ · pubmed ↗
- 2Al-Khatib S. M. Stevenson W. G. Ackerman M. J. Bryant W. J. Callans D. J. Curtis A. B. (2017). AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Circulation 138 (13), e 272–e 391. 10.1161/CIR.0000000000000549 29084731 · doi ↗ · pubmed ↗
- 3Ambry Genetics (2025). Genetic testing for long QT|inherited arrhythmia|Long QT Next and Rhythm Next. Available online at: https://www.ambrygen.com/providers/genetic-testing/14/cardiology/longqtnext-rhythmnext (Accessed July 17, 2025).
- 4Anderson C. L. Delisle B. P. Anson B. D. Kilby J. A. Will M. L. Tester D. J. (2006). Most LQT 2 mutations reduce Kv 11.1 (h ERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 113 (3), 365–373. 10.1161/CIRCULATIONAHA.105.570200 16432067 · doi ↗ · pubmed ↗
- 5Anderson C. L. Kuzmicki C. E. Childs R. R. Hintz C. J. Delisle B. P. January C. T. (2014). Large-scale mutational analysis of Kv 11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 5, 5535. 10.1038/ncomms 6535 25417810 PMC 4243539 · doi ↗ · pubmed ↗
- 6Andrássy G. Szabo A. Ferencz G. Trummer Z. Simon E. TahyÁ. (2007). Mental stress may induce QT‐Interval prolongation and t‐wave notching. Ann. Noninvasive Electrocardiol. Off. J. Int. Soc. Holter Noninvasive Electrocardiol. Inc. 12 (3), 251–259. 10.1111/j.1542-474X.2007.00169.x 17617071 PMC 6932412 · doi ↗ · pubmed ↗
- 7Berge K. E. Haugaa K. H. Früh A. Anfinsen O. G. Gjesdal K. Siem G. (2008). Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand. J. Clin. Lab. Invest 68 (5), 362–368. 10.1080/00365510701765643 18752142 · doi ↗ · pubmed ↗
- 8Berman H. Henrick K. Nakamura H. (2003). Announcing the worldwide protein data bank. Nat. Struct. Mol. Biol. 10 (12), 980. 10.1038/nsb 1203-980 14634627 · doi ↗ · pubmed ↗