The role of gut microbiota and its metabolites in preventing oncogenesis
Zehua Li, Jiaxi Yan, Ziyi Zeng, Linyong Zhao

TL;DR
This review explores how gut microbes and their byproducts can prevent cancer by supporting immune function and reducing harmful effects.
Contribution
The paper provides a comprehensive synthesis of mechanisms by which gut microbiota and metabolites prevent cancer.
Findings
Dysbiosis promotes cancer by disrupting immune and metabolic balance.
Microbial metabolites help prevent cancer through immune modulation and detoxification.
Strategies like probiotics and fecal transplants show promise for microbiome-based cancer prevention.
Abstract
The gut microbiota is increasingly recognized as a key determinant of cancer susceptibility, functioning as a dynamic interface between environmental exposures and host physiology. Dysbiosis disrupts immune homeostasis, epithelial integrity, and metabolic equilibrium, thereby fostering a microenvironment conducive to oncogenesis. Conversely, a balanced microbial ecosystem and its metabolites exert potent anti-tumor effects through immune modulation, maintenance of mucosal barrier function, and detoxification of carcinogens. This Review synthesizes emerging mechanistic insights into how commensal microbes and their metabolic products coordinate host defense pathways to suppress malignant transformation. We further discuss translational strategies—ranging from probiotics, prebiotics, and synbiotics to fecal microbiota transplantation and dietary interventions—that leverage microbiome…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
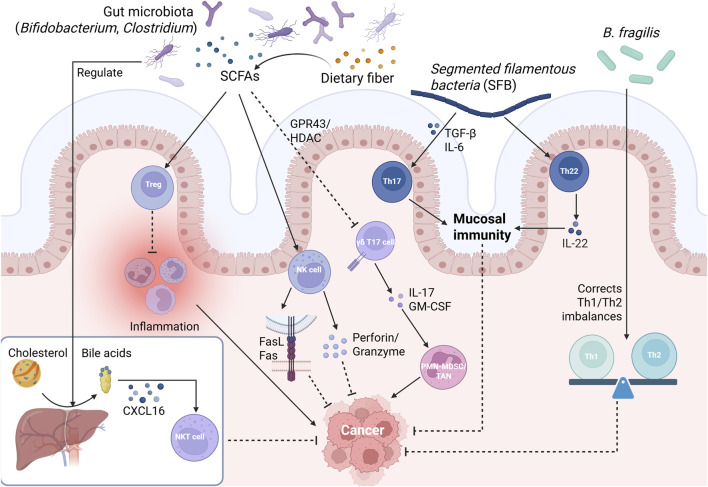 FIGURE 1
FIGURE 1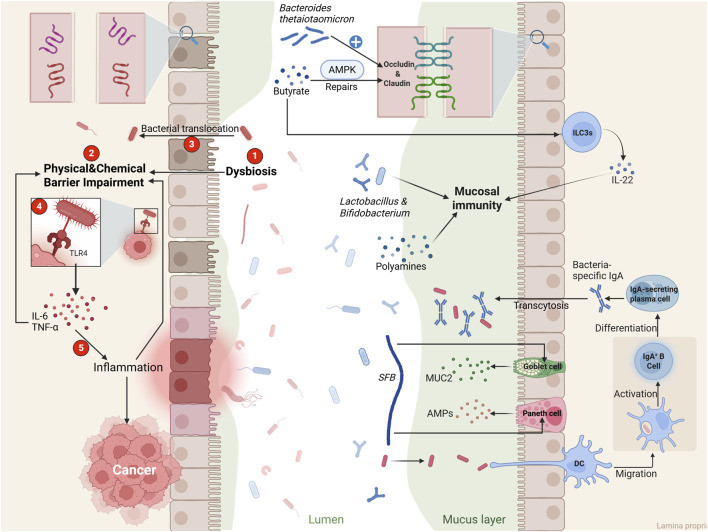 FIGURE 2
FIGURE 2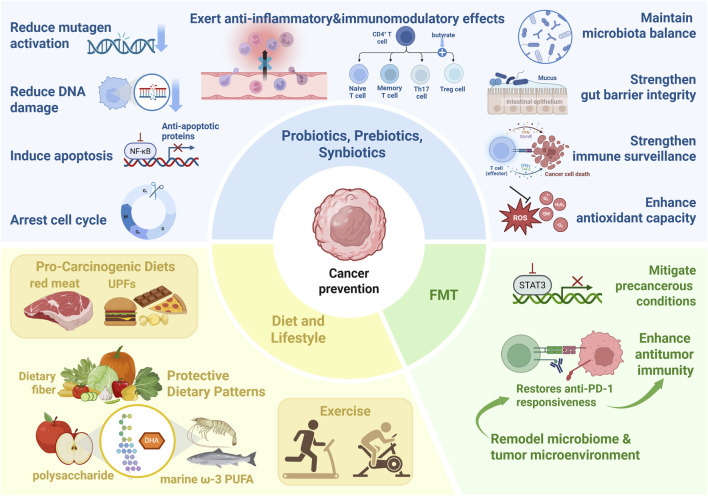 FIGURE 3
FIGURE 3| Model used | Probiotic strains used | Major findings | Mechanistic exploration | Ref | Year |
|---|---|---|---|---|---|
| DMH-induced C57BL/6 mice |
| Inhibited NF-κB; reduced mucin depletion; increased Ki-67; reduced body weight | Modulation of TLR/NF-κB pathway; altered cytokine profile |
| 2023 |
| Caco-2 cell line | Ethyl acetate extracts of | Reduced viability of Caco-2 cells (67%–71%); no effect on HUVEC cells | Activation of intrinsic apoptosis pathway; increased caspase-3 and -9 activity; downregulation of *bcl-2*, |
| 2023 |
| C57BL/6 mouse model |
| Ameliorated intestinal inflammation, tumor growth, and gut dysbiosis | Epigenetic regulation of CD8+ T cell immunity; enhanced H3K27ac binding at IL12a enhancer; reduced Saa3 expression via chromatin accessibility changes |
| 2023 |
| DMH-induced Wistar rat model | Vitamin D3; | Reduced ACF and AC counts in all treatment approaches (simultaneous, pre-, post-treatment); synergistic effect in pre-treatment; altered fecal microbiota | Modulation of Nrf2, GST, COX2, iNOS, β-catenin, PCNA expression; increased antioxidant response; anti-proliferative and anti-inflammatory effects; microbiota modulation ↑ |
| 2023 |
| AOM/DSS-induced CRC mouse model |
| Inhibited tumor formation; enhanced gut barrier; increased SCFAs; decreased LPS and pro-inflammatory cytokines | Regulated gut microbiota (↑ beneficial bacteria, e.g., |
| 2022 |
| AOM/DSS-induced CA-CRC mouse model |
| Reduced tumor number and diameter; improved gut barrier; decreased inflammation | Upregulated tight junction proteins; modulated gut microbiota (↑ beneficial bacteria, e.g., |
| 2022 |
| AOM/DSS-induced mouse model |
| Reduced inflammation, tumor number and size; improved stool consistency; increased gut microbiota diversity | Suppression of p-Akt and p-STAT3; reduction in M1/M2 macrophages; restoration of beneficial bacteria ( |
| 2021 |
| DMH-induced BALB/c mouse model |
| Reduced hepatic oxidative stress (MDA, carbonyl protein); decreased colonic IL-6, IL-17; increased acetic acid and total SCFAs; enriched Ruminiclostridium | Antioxidant activity; anti-inflammatory effects; gut microbiota modulation; SCFA production |
| 2020 |
| DMH-induced F344 rat model |
| Suppressed tumor formation; induced apoptosis and cell cycle arrest | Inhibition of AKT phosphorylation; downregulation of cyclinD1 and COX-2 |
| 2020 |
| DMH-induced rat model |
| Reduced tumor incidence (∼66%), volume, and multiplicity; prevented severe weight loss; increased apoptotic index | Upregulation of pro-apoptotic genes (Bax, Caspase-3, Caspase-9); downregulation of anti-apoptotic genes (Bcl-2, Jak-1, Akt-1); induction of apoptosis via intrinsic and extrinsic pathways; inhibition of cell proliferation |
| 2020 |
| DMH/DSS-induced rat model | Djulis + | Reduced total ACF, SIM-ACF, and MDF; downregulated PCNA and COX-2; regulated apoptosis-related proteins | Regulation of proliferative, inflammatory, and apoptotic pathways; enhanced fecal |
| 2020 |
| DMH-induced mouse model | Inulin + | Reduced CEA levels and ACF number; increased p-JNK-1; decreased β-catenin and p-GSK3β | Modulation of JNK-1/β-catenin signaling pathway; enrichment of beneficial genera ( |
| 2019 |
| DMH-induced rat model |
| Reduced tumor burden and multiplicity; upregulated Bax and p53; downregulated Bcl-2 and K-ras | Enhanced apoptosis; modulation of pro-/anti-apoptotic genes; gut microenvironment modification |
| 2018 |
| DMH-induced rat model |
| Increased antioxidant enzymes; normalized apoptosis-related protein expression | Reduction in oxidative stress; regulation of p53, Bax, Bcl-2, caspases; enhancement of antioxidant defense |
| 2018 |
| AOM/DSS-induced CRC mouse model |
| Reduced tumor incidence and proliferation; decreased histological damage | Downregulated IL-22; upregulated pro-apoptotic genes (caspase-7, caspase-9, Bik); modulated gut microbiota |
| 2017 |
| DMH-induced BALB/c mice model |
| Reduced ACF; decreased putrescine; downregulated ODC | Maintenance of polyamine metabolism; antimutagenic effect |
| 2017 |
| DMH-induced Sprague-Dawley rat model |
| Reduced tumor incidence, multiplicity, volume; induced apoptosis | Downregulation of NF-κB, COX-2, TNF-α, Bcl-2; upregulation of Bax, Casp3, p53, β-catenin |
| 2016 |
| DMH-induced rat model |
| Reduced tumor incidence, multiplicity, and volume; altered gut microbiota | Enhanced mucosal barrier (MUC2, ZO-1, occludin); increased TLR2; decreased TLR4, COX-2, caspase-3, β-catenin |
| 2016 |
|
|
| Prevented gastric inflammation and microbiota alteration induced by | Suppressed Th1/Th17 response; inhibited pro-inflammatory cytokines; modulated gastric microbiota (↑ Firmicutes/Bacteroidetes; ↓ Proteobacteria) |
| 2016 |
| ApcMin/+ mouse model | Microencapsulated | Reduced total intestinal tumor number by 44% (4.5 vs. 2.5 tumors/mouse); Reduced proliferation (Ki-67); Altered immune cell infiltration | Increased CD8+ T cells in tissue; Modulated apoptosis (cleaved caspase-3); Potential immunomodulation and anti-proliferative effects |
| 2016 |
| CT26 tumour-bearing mouse model |
| Inhibited CT26 tumor growth, prolonged survival, increased NK and CD8+ T cell infiltration in tumors | Enhanced NK and CD8+ T cell activity; promotion of Th1 response; upregulation of IFN-γ; dendritic cell maturation |
| 2015 |
| Gallus gallus (chicks) model |
| Reduced MNNG-induced genotoxicity (69%) and mutagenicity (61%) | Detoxification of MNNG to less toxic metabolites; reduction in inflammation |
| 2015 |
| DMH-induced rat model |
| Decreased cancer incidence (87.5%–25%); Counteracted DMH-induced gut microbiota dysbiosis | Increased |
| 2015 |
| LS174T cells; mouse colonic epithelium | p40 (a protein derived from | Upregulated | Activation of EGFR and Akt; mucin production via EGFR transactivation |
| 2014 |
| DMH-induced F344 rat model |
| Reduced ACF number (∼40%) and epithelial proliferation; improved microbiota structure | Increased SCFA (butyrate); decreased azoreductase activity; modulation of gut microbiota (increased |
| 2014 |
| PhIP-treated mouse model |
| Reduced PhIP-induced DNA damage in colonocytes; increased fecal lactobacilli; decreased β-glucuronidase and N-acetyl-β-glucosaminidase activities | Binding/sequestration of genotoxins; modulation of gut microbiota and enzyme activities; reduction in genotoxic metabolites |
| 2014 |
| DMH-induced Swiss mouse model |
|
| Strain-specific protection; reduction mainly in small ACF (≤3 crypts); possible modulation of microbiota or immune response |
| 2013 |
| Cyclic DSS-treated rat model | Blueberry husks + Probiotic mixture ( | Reduced DAI, number of colonic ulcers and dysplastic lesions; decreased fecal Enterobacteriaceae; increased lactobacilli; mitigated liver injury | Altered SCFA profiles (decreased butyrate, increased acetate); reduced bacterial translocation; modulation of gut microbiota |
| 2012 |
| DMH-induced ICR mouse model |
| Inhibited ACF formation; reduced colon tumor multiplicity and size | Suppressed colonic mucosa cellular proliferation; enhanced phagocytic activity of macrophages |
| 2011 |
| PhIP-induced ACF rat model; DMH-induced tumors rasH2 mice model | Yogurt ( | Reduced ACF and ACs in rats; reduced number of colorectal tumors in rasH2 mice | Possible immunomodulation; antioxidant activity; alteration of gut microbiota or mutagen sequestration |
| 2010 |
| DMH-induced rat model |
| Significantly reduced DNA damage (54.7% vs. 88.1% in DMH control) and tumor incidence in colon | Reduction in free radicals, prevention of DNA adduct formation; enhancement of antioxidant activity |
| 2009 |
| AOM-induced Fisher rat model | EPS-producing and non-EPS-producing lactic cultures (e.g., | Reduced colon tumor incidence and multiplicity with some strains; no correlation between EPS properties and chemoprevention | Reduction in COX-2 activity; possible interaction of EPS/metabolites with milk components |
| 2009 |
| WD-induced CAC mouse model | Probiotic VSL#3 + Metformin | Combination significantly ameliorated colitis and tumor growth; reduced macrophage infiltration; maintained epithelial integrity | Enhanced late apoptosis via inhibition of cyclin D1 and Bcl-2; activation of pro-apoptotic ERK and AMPK pathways |
| 2016 |
| AOM/DSS mouse model | Probiotic VSL#3 + Balsalazide | Significantly reduced total tumor number and macrophage infiltration; decreased F4/80+ macrophages in tumor stroma | Suppression of IL-6/STAT3 pathway; decreased MIP-1β, MCP-1, IL-6, IL-10; downregulation of BCL2, upregulation of BAX and p-ERK. |
| 2016 |
| DSS-induced chronic colitis mouse model | Probiotic VSL#3 | Attenuated DAI, reduced dysplasia and adenocarcinoma incidence; no carcinoma in concurrent VSL#3 group | Reduced PCNA labeling index; decreased TNF-α, IL-1β, IL-6, COX-2; increased IL-10 |
| 2015 |
| AOM/III0−/− mouse model | Probiotic VSL#3 | Did not reduce tumorigenesis or inflammation; Enhanced tumor penetrance, multiplicity, and invasion in one cohort | Altered mucosal microbiota composition (16-fold decrease in a |
| 2013 |
| AOM/DSS and H.typhlonius/IL-10−/− mouse models | Probiotic VSL#3 | Reduced disease activity, tumor number, and size; decreased adenoma/adenocarcinoma formation | Upregulation of TNF-α, PPARγ, angiotatin; downregulation of COX-2; increased IL-17+ CD4+ T cells in MLN and Tregs in lamina propria |
| 2012 |
| TNBS-induced rat model | Probiotic VSL#3 | Delayed transition from inflammation to dysplasia; reduced macroscopic damage; no carcinoma in VSL#3 group vs. 29% in controls | Increased angiotatin and VDR expression; decreased alkaline phosphatase; modulation of gut microbiota richness/diversity |
| 2011 |
| Human colon cancer cell lines | Conditioned medium from | Inhibited viability and proliferation of CRC cells; no effect on normal colon cells | Secreted proteins >100 kDa (e.g., α-mannosidase) induced apoptosis and inhibited proliferation |
| 2024 |
| CRC xenograft mouse model | α-mannosidase (from | Suppressed tumor growth in HCT116 and HT29 xenografts | Decreased Ki-67+ cells; increased TUNEL+ cells; induction of apoptosis and inhibition of proliferation |
| 2024 |
| Germ-free AOM/DSS mouse model |
| Reduced tumor number and load; increased apoptosis | Demonstrated direct antitumor effect independent of microbiota modulation |
| 2024 |
| Sprague-Dawley rat model/NU/NU nude mouse model | Microencapsulated | Targeted release in intestines; reduced tumor volume (−98.87%) and weight (−89.27%) | Upregulation of p53 and caspase-3; downregulation of COX-2, VEGF, PECAM-1; induction of apoptosis and anti-angiogenesis |
| 2022 |
| DMH-induced rat model | Isomatooligosaccharides (IMOs) | Inhibited ACF formation (29.4% reduction); reduced serum inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-8); improved gut barrier function; modulated gut microbiota | Increased abundance of |
| 2022 |
| DMH-induced Wistar rat model | Kefir milk (a probiotic fermented milk produced from kefir grains) | Reduced tumor incidence by 100% (KNL) and 71.43% (KSL); decreased IL-1β, IL-6, TNF-α, NO; increased Romboutsia | Anti-inflammatory cytokine reduction; gut microbiota modulation; increased beneficial bacteria |
| 2022 |
| DMH-induced BALB/c mouse model | Sphingomyelin (0.05%) + | Reduced number of aberrant crypt foci (ACF); improved galactose uptake in enterocyte brush-border membrane vesicles | Enhanced intestinal physiological function; possible synergy between sphingomyelin and probiotics |
| 2022 |
| AOM-induced rat model | Inulin + | Synergistic reduction in ACF, especially those with high multiplicity | Enhanced gut fermentation; production of SCFAs; modulation of bacterial enzymes |
| 2002 |
| AOM/DSS-induced mouse model |
| Alleviated colitis and tumorigenesis; improved gut microbiota; increased SCFAs; enhanced gut barrier | Inhibited TLR4/MyD88/NF-κB signaling; reduced inflammation and macrophage infiltration |
| 2021 |
| ApcMin/+ and AOM-injected mouse model |
| Reduced tumor formation; inhibited proliferation of CRC cells; induced cell cycle arrest and apoptosis | Secretion of β-galactosidase; production of galactose; activation of oxidative phosphorylation; inhibition of Hippo pathway |
| 2021 |
| DMH-induced rat model |
| Inhibited CRC cell proliferation, induced apoptosis and cell cycle arrest; reduced tumor incidence and size in DMH-induced mice | Downregulation of TLR4–MyD88–NF-κB pathway; reduction in Th2/Th17 responses; upregulation of P21; decreased inflammation |
| 2015 |
| AOM-induced Sprague-Dawley rat model |
| Synbiotic significantly reduced neoplasm incidence and multiplicity; RS alone showed a trend; probiotic alone ineffective | Increased SCFA production; reduced epithelial cell proliferation; no change in spontaneous apoptosis |
| 2010 |
| AOM-induced Sprague-Dawley rat model |
| Synbiotic combination significantly facilitated acute apoptotic response to genotoxic carcinogen in distal colon; neither probiotic nor prebiotic alone had an effect | RS acts as metabolic substrate, creating conditions for B. lactis to exert pro-apoptotic effects; increased SCFA production; no change in spontaneous apoptosis |
| 2005 |
| ApcMin/+ mouse model |
| Inhibited intestinal tumor growth and dysplasia; reduced tumor number and size | Inactivation of EGFR, HER-2, HER-3, IGF-1R signaling; inhibition of cell proliferation; induction of apoptosis |
| 2009 |
| DMH-induced F344 rat model |
| Reduced aberrant crypt foci (ACF) by 50%; Decreased leukocytic DNA damage and plasma lipid peroxidation; Increased plasma antioxidant potential | Attenuation of DMH-induced genotoxicity and oxidative stress; Enhancement of total antioxidant capacity |
| 2007 |
| DMH-induced rat model |
| Strong adherence to Caco-2 cells; Inhibited colon cancer cell growth (dose-dependent); Reduced DMH-induced aberrant crypts by 40% | High cell surface hydrophobicity and agglutination; Proposed mechanisms include antimicrobial effects and potential induction of apoptosis |
| 2006 |
| IL-10−/− mouse model |
|
| Bacterial antigen-specific CD4+ T-cell responses; differential cytokine profiles; regional immune activation |
| 2005 |
| AOM-induced F344 rat model | Inulin enriched with oligofructose (Synergy1®); | Synergy1 significantly reduced colorectal tumor number; probiotics slightly reduced malignant tumors | Increased SCFA (butyrate); reduced proliferation; altered expression of GST-P, iNOS, COX-2; increased apoptosis in normal mucosa |
| 2002 |
| ApcMin/+ mouse model | Oligofructose | Reduced colon tumors; enhanced gut-associated lymphoid tissue (GALT) | Immunomodulation; possible prebiotic effect on gut immunity |
| 2002 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Cancer Research and Treatments · Immune cells in cancer
Introduction
1
The human gut microbiota refers to the diverse community of microorganisms, including bacteria, archaea, viruses, and fungi, that reside in the gastrointestinal tract (Sender et al., 2016). This highly diverse microbial community has co-evolved with the human host over millennia, confers a broad spectrum of physiological benefits, including nutrient metabolism, immune modulation, and the maintenance of mucosal integrity (Backhed et al., 2005; Wu and Wu, 2012; Hill and Artis, 2010). Perturbations of this equilibrium, termed dysbiosis, have been increasingly implicated in the pathogenesis of a wide range of chronic and multifactorial diseases (Ivleva and Grivennikov, 2022). Over the past two decades, research on the gut microbiome has expanded substantially, driven by advances in next-generation sequencing, computational pipelines tailored to microbiome data, and experimental platforms enabling hypothesis testing at both high- and low-throughput (Bharti and Grimm, 2021; Wensel et al., 2022; Li et al., 2024). In parallel, and as a direct consequence of these developments, microbes have been increasingly recognized as pivotal mediators linking immune responses with cancer development.
Oncogenesis, the process underlying cancer development, results from the complex interplay of genetic predispositions, environmental exposures, and lifestyle factors that collectively promote uncontrolled cellular proliferation and tumor progression (Hanahan and Weinberg, 2011). Although accumulating evidence suggests that dysbiosis of the gut microbiota and its metabolites are associated with oncogenesis, significant limitations remain in its interpretation (Zhao et al., 2023). Notably, dysbiosis does not represent an absolute or universally defined state; instead, its characteristics are highly dependent on both the host and the specific disease context (Scott et al., 2019). Therefore, when analyzing the landscape of the gut and intratumoral microbiome and its metabolites, it is essential to account for inter-patient variability to elucidate the mechanisms underlying cancer initiation (Wong and Yu, 2023; El and Garrett, 2023). At the molecular level, these mechanisms are multifaceted and can be broadly categorized into genomic integration, genotoxicity, chronic inflammation, immune dysregulation and metabolic reprogramming.
The rationale for examining gut microbiota and its metabolites as a promising strategy for cancer prevention lies in its dual role as a regulator of host physiology and disease susceptibility (Gou et al., 2024; Yu et al., 2024). By maintaining immune surveillance, certain microbial environments may reduce cancer risk and prevent oncogenesis.
In this review, we examine recent studies investigating the protective roles of the gut microbiota in carcinogenesis. We highlight synergistic anti-oncogenic mechanisms driven by microbes, with particular emphasis on microbially mediated immune modulation, maintenance of epithelial barrier integrity, and detoxification of carcinogens. Specifically, we address how diverse immune cell populations interact with host microbial communities to prevent chronic inflammation, how commensal microbes preserve mucosal immune homeostasis, and how microbial metabolites contribute to cancer prevention. Furthermore, we evaluate both the potential and the limitations of microbiota- and metabolite-based interventions for personalized cancer suppression, including probiotics, prebiotics, fecal microbiota transplantation, and dietary modulation. Finally, we underscore the many unresolved aspects of microbiota research in oncogenesis and outline potential directions for advancing this rapidly evolving field.
Microbiota dysbiosis and cancer risk
2
Dysbiosis, broadly defined as an abnormal alteration in the composition or function of the gut microbiota and its metabolites, has been increasingly linked to elevated cancer risk. Rather than representing a uniform condition, dysbiosis can disrupt host–microbe interactions in ways that vary across individuals and disease contexts. Emerging evidence demonstrates that such disruptions can profoundly alter metabolic activity, immune regulation, and epithelial barrier integrity, thereby fostering a microenvironment conducive to oncogenesis (Ivleva and Grivennikov, 2022; He et al., 2025; Nobels et al., 2025; Schwabe and Jobin, 2013; Francescone et al., 2014). Here, we selected colorectal cancer (CRC), hepatocellular carcinoma (HCC), gastric cancer (GC), and breast cancer (BC) as representative malignancies because they illustrate direct, axis-mediated, pathogen-associated, and endocrine-driven mechanisms of microbiota-associated carcinogenesis.
Colorectal cancer (CRC)
2.1
Multiple studies have reported that the development of CRC is closely linked to interactions between various microbes, their metabolites, and the intestinal epithelial barrier (Herlo et al., 2024; Wu et al., 2024). Specifically, the dysbiosis can lead to a pro-oncogenic environment, resulting in chronic inflammation and immune dysregulation (Wong and Yu, 2023). Consequently, clinical cohort studies of CRC patients exhibit enrichment of pro-oncogenic taxa such as Fusobacterium nucleatum, enterotoxigenic Bacteroides fragilis, and pks ^ + ^ Escherichia coli (Wang and Fang, 2023; Sears and Pardoll, 2011; Jans and Vereecke, 2025) and depletion of beneficial gut bacteria such as Bifidobacterium, Clostridium butyricum, Faecalibacterium prausnitzii, Roseburia intestinalis, and Streptococcus thermophilus (Lopez-Siles et al., 2017; Kang et al., 2023; Correa et al., 2005). Preclinical fecal microbiota transplantation (FMT) studies have further validated the causal role of gut microbiome dysbiosis in CRC development. Germ-free (GF) mice receiving microbiota from CRC patients exhibited pronounced tumor-promoting effects, such as enhanced epithelial cell proliferation, increased polyp formation, greater dysplasia, and elevated inflammatory markers, compared with those receiving transplants from healthy donors (Wong et al., 2017). Nevertheless, the causal relationship between specific gut bacterial species and the emergence of oncogenic somatic mutations remains to be fully elucidated, although an increasing number of mechanistic studies have provided compelling evidence supporting this association.
Hepatocellular carcinoma (HCC)
2.2
Evidence from both clinical studies and preclinical animal models indicates that dysbiosis influences hepatocarcinogenesis through bidirectional communication along the gut–liver axis. In a preclinical study investigating fecal biomarkers, significant alterations in gut microbiota composition were observed during the progression of liver disease, characterized by markedly increased abundances of Bacteroides spp., Clostridium cocleatum, and Desulfovibrio spp., which correlated with elevated circulating LPS levels (Xie et al., 2016). LPS-mediated activation of TLR4 signaling in hepatocytes and Kupffer cells was shown to promote chronic hepatic injury and tumorigenesis (Jing et al., 2012). Collectively, these findings underscore the pivotal role of the gut microbiota in the progression and pathogenesis of hepatocellular carcinoma.
Bacterial metabolites and endotoxins circulating in the portal vein serve as critical mediators of hepatic inflammation, fibrosis and subsequent HCC progression, as their levels correlate with the severity of intestinal barrier disruption and microbial dysbiosis (Albillos et al., 2020; Guan et al., 2022). A compelling example is that the deficiency of the inflammasome sensor NLRP6 in mice leads to a dysbiotic gut microbiota and hepatic inflammatory microenvironment. This dysbiosis promotes hepatocellular carcinoma development by shaping the hepatic inflammatory microenvironment through activation of TLR4 and TLR9 signaling (Schneider et al., 2022). Another study reported a reduced abundance of Bacteroides thetaiotaomicron in patients with recurrent hepatocellular carcinoma (HCC). Further analysis revealed that acetic acid produced by B. thetaiotaomicron can modulate macrophage polarization toward the M1 phenotype, thereby enhancing the cytotoxic activity of CD8^+^ T cells and inhibiting HCC tumor growth (Ma et al., 2024).
Gastric cancer (GC)
2.3
It has been widely reported that Helicobacter pylori (H.pylori) infection plays a crucial role in the initial steps of carcinogenesis by exacerbate chronic inflammation and progressive degradation of the architecture and function of the gastric epithelium (Chen et al., 2013).
In general, gastric cancer–associated dysbiosis is characterized by a microbial community with genotoxic potential, marked by reduced microbial diversity, decreased Helicobacter abundance, and enrichment of newly emerging or opportunistic bacterial genera (Coker et al., 2018; Zeng et al., 2024; Lei et al., 2024). While H. pylori remains the most established microbial risk factor, non-H. pylori microbiota also play significant roles in gastric carcinogenesis (Liu et al., 2022). By promoting M2 macrophage polarization through activation of the TLR4/PI3K/Akt signaling pathway, Propionibacterium acnes may act as a potential contributor to gastric cancer progression in addition to H. pylori. A recent study further suggested that F. nucleatum infection increases the secretion of exosomes by gastric cancer (GC) cells, thereby promoting GC progression (Li et al., 2021a). Notably, although overall α-diversity decreases in carcinoma tissues, increased abundances of non-Helicobacter Proteobacteria, Firmicutes, and Actinobacteria have been detected in gastric cancer specimens (Ferreira et al., 2018). Moreover, the gastric cancer–associated microbiota exhibits enhanced nitrate and nitrite reductase activities, facilitating the formation of N-nitroso compounds—potent mutagens that accelerate gastric carcinogenesis.
In summary, while H. pylori remains the principal bacterial agent associated with gastric cancer, broader dysbiosis within the gastrointestinal microbiota also play a substantial role in GC progression.
Breast cancer (BC)
2.4
Emerging evidence indicates that the microbiota, both within the gut and the mammary gland, exerts significant influence over BC pathogenesis through metabolic, endocrine, and immunological pathways (Plaza-Diaz et al., 2019). Of these associations, the link between breast cancer and the microbial “estrobolome” has received the greatest research attention (Sui et al., 2021). It is a consortium of microbial genes responsible for estrogen metabolism (Ervin et al., 2019). Clinical observational studies and mechanistic in vitro analyses suggest that microbial imbalance, via heightened β-glucuronidase activity, enhances the reactivation of conjugated estrogens, thereby increasing systemic estrogen exposure and promoting estrogen receptor–positive tumor development (Arnone and Cook, 2022). Studies examining the relationship between gut microbial diversity and estrogen levels in postmenopausal women with BC have shown that variability in gut microbiota composition is associated with differences in estrogen metabolism and circulating estrogen concentrations. Specifically, clinical studies in postmenopausal women have shown that lower microbial diversity correlates with elevated systemic estrogen levels, suggesting that reduced gut microbial diversity may contribute to an increased risk of breast cancer (Wu et al., 2023; Flores et al., 2012).
Microbiota dysbiosis has emerged as a unifying yet context-specific factor in cancer development. Across multiple malignancies, disruptions in microbial composition and metabolism promote chronic inflammation, epithelial barrier dysfunction, and genotoxic stress. Collectively, these findings position gut microbiota imbalance as a central modulator of oncogenesis and a promising therapeutic target. Conversely, when in equilibrium, the gut microbiota plays an equally vital role in maintaining host homeostasis and preventing tumor initiation. Thus, the relationship between the microbiota and cancer is inherently dualistic: while the loss of microbial diversity and function can drive oncogenesis, a stable and metabolically active microbiome can confer protection.
Mechanisms by which gut microbiota and its metabolites prevent oncogenesis
3
Rationale for microbial focus in oncogenesis
3.1
The gut microbiota acts as a critical interface connecting environmental carcinogens and host susceptibility to cancer (Azcarate-Peril et al., 2011). Metagenomic studies show cancer patients have reduced gut microbiota diversity, especially fewer butyrate-producing bacteria, versus healthy individuals. The dysbiosis, characterized by diminished community stability, heightens exposure to carcinogens and promotes maladaptive immune responses (Bhatt et al., 2017; Garrett, 2015). Importantly, microbial metabolites play a dual role in both promoting and suppressing cancer development, positioning gut microbiota regulation as a promising strategy for cancer prevention (Bose and Mukherjee, 2019). The profound influence of gut microbiota on host metabolism highlights their potential as modifiable targets for interrupting carcinogenic pathways (Hanus et al., 2021). Specifically, the gut microbiota prevent oncogenesis through synergistic mechanisms, including modulation of the immune system, preservation of epithelial barrier integrity, and detoxification of carcinogens. Together, these processes reduce the risk of DNA damage and chronic inflammation—key drivers of tumorigenesis (Tsilimigras et al., 2017; Sun et al., 2025).
Immune system modulation
3.2
The gut microbiota regulates systemic immunity through interacting with host cells and influencing both innate and adaptive immune responses essential for protecting against precancerous lesions (Garrett, 2015). Microbial metabolites, such as short-chain fatty acids (SCFAs) and polyamines, function as immune signaling molecules that regulate inflammatory pathways, contributing to prevent chronic inflammation—a known precursor to DNA damage and cancer development (Tsilimigras et al., 2017; Yu and Zhong, 2022). Dysbiosis disrupts this immune balance, evident in the skewed Th17/Treg ratio and diminished cytotoxic activity, both are directly associated with increased cancer risk across multiple organs. Maintaining microbial balance is therefore critical for sustaining immune function and reducing cancer risk (Levy et al., 2017). This balanced immunity not only eliminates oncogenic agents but also restrains excessive inflammation, thereby blocking carcinogenic pathways.
Foundational immune programming
3.2.1
Studies in GF mouse models demonstrate that the absence of microbiota leads to underdeveloped mucosal immunity, characterized by fewer Peyer’s patches, IgA^+^ plasma cells, and CD8^+^ intraepithelial lymphocytes, weakening their ability to detect and eliminate oncogenic viruses and bacteria; these defects can be reversed by colonizing the microbiota (Chung and Kasper, 2010).
Pattern recognition receptors (PRRs) sense microbe-associated molecular patterns (MAMPs) from commensals, shaping host immune responses (Chu and Mazmanian, 2013). Early exposure to microbiota adjusts mononuclear phagocytes through chromatin remodeling, enhancing systemic antiviral and antibacterial immunity to eliminate potential tumor cells (Ganal et al., 2012; Takiishi et al., 2017). Type I interferons (IFN-I) and NK cells are central to antiviral immunity: IFN-I activates DCs to promote NK and CD8^+^ T cell activity, while NK cells eliminate MHC-I-deficient tumor cells via perforin/granzyme or Fas/FasL pathways (Kawai and Akira, 2006; Tougeron et al., 2013). Commensal microbiotas prime these responses—critical since ∼10% of cancers (e.g., HPV-induced cervical cancer, HBV-induced HCC) stem from viral infections, and robust antiviral immunity clears infected cells before malignant transformation (Schiller and Lowy, 2021; Plummer et al., 2016; Winkler and Thackray, 2019).
This early immune programming lays the foundation for long-term immune competence, helping to prevent chronic inflammation that fosters the formation of a tumor-promoting microenvironment.
Anti-tumor immunity enhancement
3.2.2
Treg cells
3.2.2.1
Regulatory T cells (Tregs) suppress excessive inflammation and maintain immune tolerance, thereby inhibiting chronic inflammation-driven tumorigenesis (Levy et al., 2017; Arpaia et al., 2013). Studies show that Bifidobacterium infantis expands CD4^+^CD25^+^Foxp3^+^ Tregs to suppress inflammation-related cancer (O'Mahony et al., 2008). Indigenous Clostridium species (clusters IV/XIVa) expand colonic Tregs by inducing TGF-β activation and promoting IL-10^+^CTLA4^high^ iTreg differentiation, which protects against colitis and systemic IgE responses (Atarashi et al., 2011). Microbial-derived SCFAs (acetate/propionate/butyrate) further enhance Treg numbers, function, and Foxp3/IL-10 expression via GPR43 (Ffar2)-dependent HDAC inhibition, restoring colonic Treg homeostasis and ameliorating inflammation in mice (Smith et al., 2013). Conversly, Dysbiosis disrupts this Treg-Th17 balance, enabling inflammation and tumor progression (Zhang et al., 2019).
γδ T cells
3.2.2.2
γδ T cells contribute to tumor prevention through direct cytotoxicity (granzyme/perforin) and IFN-γ-mediated antitumor activity (Hannani et al., 2012; Kong et al., 2009). However, IL-17-producing γδ T cells (γδT17) promote tumor progression by driving immunosuppressive PMN-MDSC and TAN accumulation via IL-17/GM-CSF secretion (Wu et al., 2014; Coffelt et al., 2015). Commensal bacteria maintain protective γδ intraepithelial lymphocytes (IELs) and critically suppress γδT17 activity via metabolites like SCFAs (Li et al., 2017; Wu et al., 2017; Dupraz et al., 2021). Conversely, microbial dysbiosis impairs barrier function, reduces SCFAs (alleviating γδT17 suppression), and promotes γδT17 polarization, ultimately enabling immunosuppression and tumor development (Jenkins et al., 2019).
Th17 cells
3.2.2.3
Segmented filamentous bacteria (SFB) specifically induce Th17 cells, enhancing mucosal defense against pathogens (Ivanov et al., 2009). Commensal microbiota collectively modulate Th17 cell differentiation via TGF-β/IL-6 signaling (Chung and Kasper, 2010; Zhang et al., 2019).
Th22 cells
3.2.2.4
SFB induce Th22 cell differentiation independent of classical Th17 cells and TGF-β signaling. Experimental results demonstrate that SFB colonization significantly enhances IL-22^+^CD4^+^ T cell numbers during infection, confirming that specific gut microbiota modulate Th22 development to strengthen mucosal defense. Th22 cells may reduce cancer risk by secreting IL-22 to enhance intestinal barrier integrity and suppress chronic inflammation (Roy et al., 2021).
Th1/Th2 balance
3.2.2.5
Bacteroides fragilis monocolonization corrects Th1/Th2 imbalances in GF mice. By maintaining T-cell homeostasis and suppressing pathological inflammation, these microbiota-mediated immunomodulatory effects collectively reduce risks of inflammation-driven carcinogenesis (Zhang et al., 2019).
Natural killer (NK) cells
3.2.2.6
Natural killer (NK) cells directly recognize and eliminate tumor cells, playing a critical role in antitumor immunity. In vitro studies have shown that gut microbiota-derived short-chain fatty acids (SCFAs) enhance NK cell cytotoxicity against tumors like multiple myeloma by reducing anti-inflammatory IL-10 secretion and promoting extracellular vesicle release (Perez et al., 2024). Conversely, intestinal microbiota suppresses intratumoral NK cell infiltration and IFNγ expression in pancreatic cancer, accelerating tumor progression (Yu et al., 2022).
Natural killer T (NKT) cells
3.2.2.7
NKT cells are essential for liver antitumor surveillance against primary and metastatic tumors. Gut microbiome-regulated bile acid metabolism controls hepatic NKT cell accumulation via the CXCL16-CXCR6 axis: primary bile acids (e.g., CDCA) induce CXCL16 on liver sinusoidal endothelial cells, promoting NKT cell recruitment and tumor suppression, while secondary bile acids (e.g., ω-MCA) inhibit this pathway (Ma et al., 2018) (Figure 1).
The Interplay Between the Gut Microbiome, Its Metabolites, and Immune Cells. Commensal microbiota (Bifidobacterium, Clostridium) produce short-chain fatty acids (SCFAs) that exert coordinated immunoregulatory effects by: Sender et al. (2016) expanding Treg cells via GPR43-mediated HDAC inhibition to suppress inflammation; Backhed et al. (2005) inhibiting the pro-tumor γδT17-PMN-MDSC/TAN axis; Wu and Wu (2012) enhancing NK cell cytotoxicity. Concurrently, segmented filamentous bacteria (SFB) induce Th17 cell differentiation via TGF-β/IL-6 signaling and drive Th22 cell-mediated IL-22 secretion, thereby reinforcing mucosal immunity. Separately, microbiome-processed primary bile acids (e.g., CDCA) activate the CXCL16-CXCR6 axis to recruit hepatic NKT cells for tumor surveillance. Additionally, Bacteroides fragilis monocolonization corrects Th1/Th2 imbalances in germ-free hosts to reduce carcinogenesis risk. Collectively, these pathways orchestrate anti-tumor immunity by preventing chronic inflammation, preserving epithelial barrier integrity, and enabling direct elimination of transformed cells. Notes: Solid arrows (→) indicate activation or induction of the immediate downstream process. Blunted lines (---) indicate inhibition or suppression of the immediate downstream target.
Maintenance of epithelial barrier integrity and mucosal immunity
3.3
The intestinal epithelial barrier, consisting of physical, chemical, and immunological components, prevents harmful substances, such as carcinogens and pathogenic bacteria, from entering the body (Bose and Mukherjee, 2019). Disruption of this barrier facilitates bacterial translocation and triggers systemic inflammation, which can induce DNA damage and promote cancer development (Schwabe and Jobin, 2013; Yu and Zhong, 2022). The gut microbiota and its metabolites help maintain the integrity of the epithelial barrier and mucosal immunity, thereby lowering cancer risk. Dysbiosis-induced barrier dysfunction, which increases carcinogen exposure and amplifies inflammatory responses, underscores the importance of maintaining microbial homeostasis for cancer prevention.
Intestinal barrier formation and maintenance
3.3.1
The barrier regulates luminal factor-immune interactions; disruptions of this barrier lead to “leaky gut,” which has been linked to IBD, metabolic diseases, and colorectal cancer (Neurath et al., 2025).
Commensals fortify the physical barrier: The physical barrier is formed by intestinal epithelial cells with tight junctions, controlling permeability and preventing microbial translocation. Bacteroides thetaiotaomicron upregulates tight junction proteins (claudin, occludin), while butyrate activates AMPK to repair tight junctions (Takiishi et al., 2017; Miao et al., 2016; Groschwitz and Hogan, 2009). Besides, Impaired intestinal barrier function permits microbiota access to epithelial TLRs, activating calcineurin-NFATc3 signaling. This pathway promotes cancer stem cell survival and proliferation, driving tumor development. Inhibiting microbiota-TLR interactions or blocking calcineurin-NFATc3 activation suppresses tumorigenesis, highlighting barrier integrity as a critical preventive mechanism (Peuker et al., 2016).
Chemical barrier maintenance: The chemical barrier comprises mucins, antimicrobial peptides, and secretory IgA, which inhibit microbes through secretion and immune exclusion. Microbialstimuli (e.g., SFB) induce mucin (MUC2) and AMP production; Lactobacillus and Bifidobacterium increase mucus viscosity to block carcinogens (Bhatt et al., 2017; Johansson et al., 2014). Gut microbiota-produced polyamines (e.g., putrescine, spermidine) enhance intestinal barrier integrity and suppress procarcinogenic inflammation, thereby inhibiting tumor initiation (Murray Stewart et al., 2018; Ramos-Molina et al., 2019).
Mucosal immunity
3.3.2
The maintenance of spatial homeostasis within the gut mucosal immune barrier prevents bacterial penetration and aberrant inflammatory responses. Key cellular and molecular players contribute to this homeostasis:
Cellular Effectors: Dendritic cells in the lamina propria sample luminal antigens to coordinate IgA secretion, thereby restricting bacterial translocation and maintaining barrier integrity (Gorski and Weber-Dabrowska, 2005). Concurrently, butyrate promotes the production of IL-22 by group 3 innate lymphoid cells (ILC3s), enhancing mucosal defense and epithelial repair.
Cytokine Regulation and Microbiota Control: Study indicates that IL-33 gene deficiency is associated with an increased risk of cancer; this susceptibility arises from the resultant dysbiotic microbiota (characterized by expansion of mucolytic bacteria like Akkermansia muciniphila), which promotes epithelial damage, early release of proinflammatory IL-1α, and ultimately drives colitis and tumorigenesis (Malik et al., 2016). Lipocalin 2 (LCN2), induced by the microbiota via MYD88, further reinforces this barrier by limiting bacterial iron acquisition and preventing the formation of colitogenic microbiota, thus reducing colorectal cancer risk (Singh et al., 2016; Moschen et al., 2016).
Avoiding Barrier Dysregulation: Conversely, excessive TLR4 expression, as seen in inflammatory bowel disease (IBD) or colitis-associated cancer (CAC), disrupts barrier function and fosters a proinflammatory microbiota, exacerbating colitis and tumorigenesis (Yu and Zhong, 2022; Dheer et al., 2016).
Disruption of this immune barrier homeostasis has dire consequences. When the spatial integrity is compromised (e.g., due to pathogens like H. pylori), bacterial penetration occurs. This triggers the release of potent proinflammatory cytokines (e.g., IL-6, TNF-α) and causes chronic mucosal damage. This persistent inflammation is a key oncogenic driver: it directly mediates cellular damage and proliferation conducive to cancer, and establishes a microenvironment of chronic damage (e.g., *H. pylori-*induced gastritis, a recognized precancerous lesion), significantly increasing the risk of malignancies like gastric adenocarcinoma (Fox and Wang, 2007).
Therefore, the gut microbiota’s role in actively preserving the spatial homeostasis of the immune barrier is fundamental to cancer prevention. By preventing bacterial encroachment and the ensuing cascade of abnormal inflammation and chronic tissue damage, this equilibrium significantly reduces cancer risk (Figure 2).
The Distinct Roles of Gut Microbial Homeostasis and Dysbiosis in Regulating Epithelial Barrier Integrity and Mucosal Immunity. This schematic contrasts the consequences of a balanced versus dysregulated gut microbiota. Beneficial commensal bacteria are depicted in blue, and potentially harmful bacteria in red. The right panel (“Normal condition”) illustrates a healthy microbiota-dominated state that maintains homeostasis. The left panel (“Dysbiotic condition”) depicts microbial imbalance characterized by the overgrowth of harmful bacteria, which initiates a pathological cascade. Under physiological conditions, a balanced microbiota and its metabolites (e.g., butyrate, polyamines) fortify the epithelial barrier by upregulating tight junction proteins, activating AMPK activation, and enhancing mucus production. Dendritic cell (DC)-coordinated secretion of secretory IgA (sIgA) limits bacterial translocation, while butyrate-induced IL-22 production from group 3 innate lymphoid cells (ILC3s) promotes mucosal defense and repair. Conversely, dysbiosis initiates a vicious cycle. It impairs barrier function through tight junctions disassembly and mucolysis, allowing bacterial translocation. This leads to TLR4 hyperactivation, chronic inflammation, and progressive barrier damage, collectively driving tumorigenesis.
Detoxification and metabolism of carcinogens
3.4
The gut microbiota participates in the metabolism and detoxification of carcinogens, thereby influencing cancer development (Sun et al., 2025; Louis et al., 2014). By producing specific enzymes, gut bacteria can transform carcinogens into less harmful substances or promote their excretion, reducing the damage to host cells. Conversely, dysbiosis may impair these detoxification processes, enhauncing carcinogen activity and increasing cancer risk. Therefore, maintaining gut microbiota’s normal detoxification and metabolic function is an important way to prevent cancer.
Direct detoxification
3.4.1
Gut microbial metabolism of carcinogens can have dual effects, either mitigating their harmful impact or enhancing their carcinogenicity. Normally, certain bacteria participate in carcinogen degradation. For instance, Klebsiella species deaminate melamine; some E. coli strains hydrolyze IQ-glucuronide, and gut microbes reduce azo compounds (Koppel et al., 2017; Zheng et al., 2013; Ervin and Redinbo, 2020). Microbial β-glucuronidases can decompose conjugated nitrosamines (such as BBN/EHBN), promoting their excretion in feces and reducing the risk of bladder cancer (Roje et al., 2024). Dysbiosis disrupts normal metabolism, leading to the accumulation of mutagens—such as altered azo reduction that enhances benzidine induced carcinogenesis (Rafii and Cerniglia, 1995).
Metabolite-mediated regulation
3.4.2
SCFAs: Produced by bacterial fermentation of fiber, SCFAs inhibit HDAC to suppress colorectal cancer cell proliferation (Li et al., 2018). They activate GPR43 to secrete IL-18 and AMPs, enhancing barrier function. However, butyrate may promote metastasis in ATP-depleted tumors via β-oxidation (Donohoe et al., 2014; Vipperla and O'Keefe, 2016).
Polyamines: Spermine, spermidine, and putrescine (from amino acid metabolism) support epithelial turnover and lymphocyte activity but can induce DNA damage at high concentrations (Hasko et al., 2000; Pegg, 2013).
Indole-3-lactic acid: A metabolite of Lactobacillus plantarum L168, it enhances IL12a production in DCs to prime CD8^+^ T cell immunity against tumors and inhibits Saa3 to improve tumor-infiltrating CD8^+^ T cell function (Zhang et al., 2023).
Bile acid homeostasis
3.4.3
Primary bile acids, synthesized from cholesterol in the liver (e.g., cholic acid, chenodeoxycholic acid), undergo microbial metabolism in the gut—primarily via 7α-dehydroxylase produced by bacteria like Clostridium scindens—to form secondary bile acids (e.g., deoxycholic acid [DCA], lithocholic acid [LCA]). This process plays a critical role in regulating host-microbe crosstalk and carcinogenesis (Jia et al., 2018; Collins et al., 2023; Yu et al., 2020). Primary bile acids activate farnesoid X receptor (FXR), maintaining intestinal barrier integrity, inhibiting IL-17-driven inflammation, and regulating bile acid synthesis through negative feedback via fibroblast growth factor 19 (FGF19). FXR knockout mice develop spontaneous liver tumors and show increased colorectal cancer susceptibility, highlighting its tumor-suppressive role (Vavassori et al., 2009; Maran et al., 2009). Secondary bile acids like LCA also activate G-protein-coupled bile acid receptor 1 (TGR5), stimulating anti-inflammatory pathways in macrophages and enhancing energy metabolism to limit carcinogenic microenvironments (Kawamata et al., 2003; Fiorucci and Distrutti, 2015). However, high-fat diets enrich 7α-dehydroxylase-producing bacteria, increasing DCA levels—DCA induces reactive oxygen/nitrogen species (ROS/RNS) to cause DNA damage, provokes senescence-associated secretory phenotype (SASP) in hepatic stellate cells (releasing IL-6 and TNF-α), and promotes obesity-associated hepatocellular carcinoma (HCC) (Payne et al., 2008). Additionally, microbiota-derived bile acids act as androgen receptor (AR) antagonists, promoting stem-like properties of CD8^+^ T cells via CD8^+^ T cell-intrinsic AR signaling to inhibit tumor progression and potentiate anti-PD-1 therapy efficacy (Jin et al., 2025). This bidirectional interplay underscores the critical role of microbial regulation of bile acid metabolism in cancer prevention.
Therapeutic and preventive potential
4
The gut microbiota constitutes a pivotal mediator between environmental exposure and host physiology. Emerging evidence indicates that its dysregulation contributes to oncogenesis and tumor development, thereby positioning the gut microbiota as a promising target for therapeutic and preventive cancer interventions. Strategies aimed at modulating microbial composition or regulating metabolite production hold potential to suppress carcinogenesis by enhancing anti-tumor immunity and preserving both intestinal and systemic homeostasis (Gerner and Meyskens, 2004). These insights provide a rationale for the clinical translation of microbiota-targeted approaches, including probiotics and prebiotics, fecal microbiota transplantation, and dietary interventions, as complementary strategies in cancer prevention (Figure 3).
Major Microbiota-Based Approaches to Prevent Oncogenesis. The figure illustrates three major microbiota-targeted intervention approaches. Probiotics, Prebiotics, and Synbiotics exert direct anti-tumor effects (e.g., induction of apoptosis, cell cycle arrest), promote carcinogen detoxification (e.g., reduced DNA damage, inhibition of mutagen activation), modulate immune responses, and strengthen gut barrier integrity. Fecal Microbiota Transplantation (FMT) remodels the host microbiome to mitigate precancerous conditions (e.g., in IBD) and enhances antitumor immunity by restoring responsiveness to immunotherapy. Diet and Lifestyle Interventions highlight the contrasting roles of pro-carcinogenic diets (e.g., red meat, UPFs) versus protective factors (e.g., dietary fiber, omega-3 PUFAs, exercise), acting primarily through modulation of microbial composition and production of protective metabolites like SCFAs. Collectively, these strategies underscore the gut microbiota’s pivotal role as a therapeutic target in cancer prevention and treatment.
Probiotics, prebiotics and synbiotics
4.1
Probiotics are defined as “live microorganisms that, when administered in adequate amounts, confer a health benefit on the host” (Kvakova et al., 2022). A growing body of preclinical evidence supports their potential in cancer prevention through multiple mechanisms, including immunomodulation, carcinogen detoxification, direct antitumor signaling, and microbiota modulation. Prebiotics are “selectively fermented ingredients that allow specific changes in gut microbiota composition/activity”, and their combination with probiotics as synbiotics often yields enhanced efficacy (Bruno-Barcena and Azcarate-Peril, 2015).
Anti-inflammatory and immunomodulatory actions
4.1.1
Probiotics exert notable anti-inflammatory and immunomodulatory effects. Strains such as Lactobacillus rhamnosus GG-derived protein p40 activate EGFR signaling, enhance mucin production, and suppress NF-κB-mediated inflammation, thereby interrupting the inflammation-dysplasia-carcinoma sequence (Wang L. et al., 2014). Lactobacillus gasseri augments macrophage phagocytosis and promotes S-phase proliferation in RAW264.7 cells via upregulation of PCNA and cyclin A, strengthening immune surveillance (Foo et al., 2011). Lactobacillus plantarum ZDY 2013 suppresses Th1/Th17 responses and inhibits pro-inflammatory cytokines in H. pylori-infected models, preventing gastric inflammation and microbiota disruption (Pan et al., 2016). In Apc^Min/+^ mice, microencapsulated Lactobacillus acidophilus delivered in yogurt increased CD8^+^ T cell infiltration and cleaved caspase-3, indicating enhanced antitumor immunity and apoptosis (Urbanska et al., 2016). Similarly, Lactobacillus plantarum stimulated NK and CD8^+^ T cell activity, promoted Th1 response, and upregulated IFN-γ in CT26 murine tumor models (Hu et al., 2015). Certain probiotic mixtures, such as VSL#3, attenuated colitis and tumorigenesis in multiple models by reducing pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) and increasing IL-10 (Talero et al., 2015; Bassaganya-Riera et al., 2012).
Prebiotics such as inulin are fermented by beneficial bacteria like Bifidobacterium to produce butyrate, which inhibits histone deacetylases (HDACs) and promotes regulatory T cell (Treg) differentiation, contributing to anti-inflammatory and immunoregulatory effects (Smith et al., 2013; Pool-Zobel et al., 2002).
Synbiotic combinations, including inulin with L. rhamnosus and B. lactis, further potentiate these benefits by synergistically reducing tumor incidence and suppressing pro-inflammatory mediators, including COX-2 (Femia et al., 2002).
Carcinogen detoxification
4.1.2
Probiotics contribute to chemoprevention by detoxifying dietary carcinogens. Lactobacillus rhamnosus Vc directly binds the genotoxic compound MNNG, facilitating its biotransformation into non-mutagenic metabolites (Pithva et al., 2015). L. rhamnosus IMC501 reduces PhIP-induced DNA damage in colonocytes, decreases fecal β-glucuronidase and N-acetyl-β-glucosaminidase activities, and modulates microbial enzyme profiles to lower genotoxic metabolite production (Dominici et al., 2014). Probiotic consortia also downregulate procarcinogenic enzymes, including β-glucuronidase and nitroreductase in the colon lumen, thereby reducing mutagen activation (Verma and Shukla, 2013). Moreover, L. acidophilus, L. casei, and L. lactis significantly decreased DNA damage and tumor incidence in a DMH-induced rat model, likely through free radical reduction and prevention of DNA adduct formation (Kumar et al., 2010).
Prebiotics such as isomatooligosaccharides (IMOs) reduce fecal β-glucuronidase and β-glucosidase activities, decrease bile acids and amino acids, and inhibit aberrant crypt foci (ACF) formation, demonstrating protective effects against carcinogen-induced tumorigenesis (Chen et al., 2022).
Direct anti-tumor signaling
4.1.3
Several probiotics directly inhibit tumor growth through induction of apoptosis and cell cycle arrest. Streptococcus thermophilus-secreted β-galactosidase induces cell cycle arrest in colorectal cancer (CRC) cells via galactose-mediated OXPHOS activation and Hippo pathway suppression (Li et al., 2021b). Lactococcus lactis HkyuLL 10 secretes α-mannosidase, which triggers mitochondrial apoptosis via cytochrome C release and ER stress through CHOP upregulation in neoplastic cells (Su et al., 2024). Lactobacillus salivarius Ren inhibits AKT phosphorylation and downregulates cyclin D1 and COX-2, thereby inducing apoptosis and cell cycle arrest (Dong et al., 2020). Lactobacillus paracasei X12 upregulates pro-apoptotic genes (Bax, Caspase-3, Caspase-9) while downregulates anti-apoptotic genes (Bcl-2, Jak-1, Akt-1), activating both intrinsic and extrinsic apoptotic pathways (Mousavi Jam et al., 2020). Bacillus polyfermenticus exhibits strong adherence to intestinal cells and inhibits colon cancer growth in a dose-dependent manner, likely through induction of apoptosis (Lee et al., 2007).
Prebiotics such as oligofructose also reduce colon tumors and enhance gut-associated lymphoid tissue (GALT), suggesting their immunomodulatory role in tumor prevention (Pool-Zobel et al., 2002).
Synbiotics demonstrate enhanced efficacy in inducing apoptosis and suppressing tumorigenesis. For instance, resistant starch combined with Bifidobacterium lactis markedly facilitated the acute apoptotic response to genotoxic carcinogens in the distal colon, whereas neither the probiotic nor prebiotic alone was effective (Le Leu et al., 2005). In AOM-induced rats, this synbiotic combination significantly reduced neoplasm incidence and multiplicity, accompanied by increased SCFA production and reduced epithelial cell proliferation, though without altering spontaneous apoptosis (Le Leu et al., 2010). Likewise, formulation such as inulin with L. rhamnosus GG and B. lactis Bb12, significantly reduced colorectal tumor numbers and malignant progression, while increasing SCFA levels and apoptosis in normal mucosa (Femia et al., 2002).
Antioxidant effects and microbiota modulation
4.1.4
Probiotics also demonstrate antioxidant properties and modulate gut microbial composition. Lactobacillus paracasei DTA81 reduces hepatic oxidative stress (MDA, carbonyl protein) and enriches SCFA-producing Ruminiclostridium in DMH-induced mice (da Silva Duarte et al., 2020). Bacillus polyfermenticus attenuates DMH-induced genotoxicity and oxidative stress while enhancing plasma antioxidant capacity (Park et al., 2007). L. salivarius counteracts DMH-induced dysbiosis by increasing Prevotella and decreasing Ruminococcus and Bacteroides dorei, thereby restoring a healthier microbial community (Zhang et al., 2015). Lactobacillus casei ATCC 393 reduces aberrant crypt foci (ACF) and downregulates ornithine decarboxylase (ODC), helping regulate polyamine metabolism and exerting antimutagenic effects (Irecta-Najera et al., 2017).
Prebiotics such as dietary fiber and specific plant polysaccharides enrich beneficial bacteria including Lactobacillus, Bifidobacterium, Roseburia, and Eubacterium, enhancing SCFA production and strengthening gut barrier integrity (So et al., 2018; Guo et al., 2021).
Synbiotic formulations, including L. acidophilus with oligofructose/maltodextrin, alter gut microbiota, enhance mucosal barrier function (MUC2, ZO-1, occludin), and decrease pro-carcinogenic signaling pathways (TLR4, COX-2, caspase-3, β-catenin) (Kuugbee et al., 2016).
Fecal microbiota transplantation (FMT)
4.2
Fecal microbiota transplantation (FMT) is defined as the transplantation of gut microbiota from healthy donors to recipients via the upper or lower gastrointestinal route, with the aim of restoring microbial diversity and function (Chen et al., 2019). It represents an emerging therapeutic approach for cancer prevention by modulating gut microbial communities to suppress inflammatory, proliferative, and pro-carcinogenic pathways, as well as reducing microbiota-induced genotoxicity.
Currently, FMT is most extensively applied in the treatment of recurrent Clostridium difficile infection, with clinical resolution achieved in up to 81% of patients after a single infusion (van Nood et al., 2013). Furthermore, chronic inflammation and barrier disruption in IBD, driven by dysbiosis and pathogens including enterotoxigenic B. fragilis (ETBF), which activates Stat3 and promotes Th17-dependent carcinogenesis, significantly increase CRC risk (Wu et al., 2009; Grivennikov et al., 2012). In this context, FMT has been shown to induce clinical remission in 63% of IBD patients and facilitate discontinuation of immunosuppressants in 76%, thereby mitigating a key precancerous condition (Anderson et al., 2012; Wang ZK. et al., 2014). Additionally, FMT is being investigated as an adjunct to immunotherapy. Early-phase trials have demonstrated that it can restore anti-PD-1 responsiveness in refractory melanoma patients via remodeling both the gut microbiome and tumor microenvironment, highlighting its ability to enhance antitumor immunity (Baruch et al., 2021).
Looking forward, the development of lyophilized microbial consortia (e.g., F. prausnitzii-enriched capsules) offers a safer, a standardized,and more practical alternative to fresh stool for broader preventive application. However, several challenges remain, including limited long-term safety data and the risk of adverse events ranging from abdominal discomfort to serious infections or disease relapse. These concerns underscore the need for rigorous donor screening, standardized protocols, and well-controlled clinical trials before widely implemented in cancer prevention (Chen et al., 2019; Kazmierczak-Siedlecka et al., 2020).
Diet and Lifestyle Interventions
4.3
An accumulating body of evidence underscores the critical role of diet and lifestyle factors in directly shaping the composition and functional capacity of gut microbial ecosystems. The gut microbiome integrates these environmental cues with host physiology and metabolism, thereby modulating key biological processes involved in tumor initiation and progression (Song and Chan, 2019). In terms of dietary patterns, distinct nutritional profiles exert opposing effects on carcinogenesis, largely mediated through microbial modulation.
Pro-carcinogenic diets
4.3.1
The Western dietary pattern, characterized by high consumption of saturated fats, refined sugars, and processed meats along with inadequate fiber intake, has emerged as a major risk factor for cancer development (Cordain et al., 2005). Increasing quantitative studies have demonstrated that continuous overconsumption of red meat and ultra-processed foods (UPFs) constitutes a key drivers of various tumor types (Ibragimova et al., 2021). Regular intake of red and processed meat, defined as unprocessed mammalian muscle such as beef and lamb along with chemically preserved products like bacon and salami, generates heterocyclic amines during high-temperature cooking that form mutagenic DNA adducts (Carr et al., 2017). Consistent findings from prospective epidemiological studies and meta-analyses show that red and processed meat convincingly elevates CRC risk by 20%–30% (Aykan, 2015). Concurrently, UPFs, which are industrial formulations containing five or more synthetic additives such as emulsifiers, artificial sweeteners, and hydrogenated fats, have been linked to increased cancer risk. A recent cohort study in the French population reported that a 10% increase indietary UPFs proportion was associated with an over 10% increase in the risk of overall cancer and breast cancer (Fiolet et al., 2018). Furthermore, high UPF intake has been positively linked to colorectal adenomas, particularly advanced and proximal lesions, with a pronounced dose-dependent effect observed among smokers (Fliss-Isakov et al., 2020).
Protective dietary patterns
4.3.2
In contrast, several protective dietary patterns modulate the gut microbiota to suppress tumorigenesis.
Dietary fiber is a major substrate for microbial fermentation, producing beneficial short-chain fatty acids (SCFAs) such as butyrate, acetate, and propionate (So et al., 2018). Higher fiber intake enriches beneficial bacteria including Lactobacillus spp. and butyrate producing genera such as Bifidobacterium, Roseburia, and Eubacterium, thereby enhancing SCFA production (Holscher et al., 2015). Crucially, dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner, primarily through histone deacetylase inhibition. This protective effect requires both fiber and butyrate-producing bacteria, as demonstrated by the synergistic reduction in tumor incidence when a high-fiber diet is combined with Butyrivibrio fibrisolvens supplementation reducing (Donohoe et al., 2014). Beyond direct epigenetic modulation, SCFAs contribute to immune regulation by promoting the differentiation and function of regulatory T cells (Treg), thereby suppressing inflammation and enhancing antitumor immunity. Butyrate and propionate induce Treg generation through histone deacetylase inhibition and GPR109a activation (Singh et al., 2014). The presence of SCFA-producing bacteria is linked to improved responses to immunotherapy, underscoring the role of fiber and microbial metabolites in modulating tumor immunity (Matson et al., 2018; Gopalakrishnan et al., 2018). Collectively, these findings demonstrate that dietary fiber influences colorectal cancer prevention through microbial production of SCFAs and immunomodulatory effects.
Beyond fiber, specific plant-derived polysaccharides from sources such as Ganoderma lucidum, Zizyphus jujuba, apple, and Dendrobium officinale have demonstrated profound anti-tumor effects. Mechanistically, these polysaccharides restore microbial balance and enhance SCFA production (Ji et al., 2019; Li et al., 2020), strengthen gut barrier integrity (Guo et al., 2021; Liang et al., 2019), inhibit pro-carcinogenic signaling pathways like Wnt/β-catenin, and potentiate anti-tumor immunity by improving the metabolic fitness while reducing exhaustion of cytotoxic T cells within the tumor microenvironment. This collective evidence highlights the role of these polysaccharides in modulating the gut microbiota-metabolite-immune axis for cancer prevention.
Marine omega-3 polyunsaturated fatty acids (PUFAs) also exhibit protective effects. Meta-analysis of biospecimen studies indicates that higher tissue levels of long-chain n-3 PUFAs, particularly EPA and DHA, are associated with a reduced risk of colorectal cancer (Yang et al., 2014). A large prospective cohort study further suggested a potential complex relationship with CRC subtypes and a possible latency period for the protective effect in men (Song et al., 2014). Importantly, higher intake of marine ω-3 PUFAs following CRC diagnosis has been associated with lower CRC-specific mortality, with incremental increases conferring additional survival benefits (Song et al., 2017). The anti-tumor mechanisms may extend beyond anti-inflammation; for example, a clinical intervention study using EPA-free fatty acid in CRC patients with liver metastases demonstrated anti-angiogenic activity in tumor tissue and a remarkable overall survival benefit despite similar early recurrence rates (Cockbain et al., 2014).
Physical exercise
4.3.3
Exercise increases gut microbiota diversity (e.g., Faecalibacterium, Akkermansia) and enriches butyrate-producing taxa (e.g., Roseburia, Eubacterium) (Clarke et al., 2014; Bressa et al., 2017). This elevates SCFA production, particularly butyrate (Allen et al., 2018), which suppresses inflammation via HDAC inhibition and Treg differentiation, thereby reducing pro-inflammatory cytokines such as IL-17 (Levy et al., 2017; Zhang et al., 2019). Concurrently, butyrate strengthens gut barrier integrity via AMPK-dependent tight junction repair and mucin synthesis (Takiishi et al., 2017), while also promoting anticancer immunity through stimulating cytotoxic T-cell activation and inducing apoptosis in transformed cells. Collectively, these microbial and immunometabolic shifts contribute to reduced cancer risk.
Conclusion and future perspectives
5
As the ancient wisdom reminds us, “to prevent is better than to cure”. This review has synthesized compelling evidence that the gut microbiota stands as a pivotal modifier of cancer risk, orchestrating its effects through multifaceted mechanisms including modulation of the immune system, maintenance of epithelial barrier integrity, and detoxification of carcinogens (Bhatt et al., 2017). These common findings emphasize that preserving the probiotic nature of the symbiotic microbial community is fundamental to suppressing chronic inflammation, neutralizing genotoxic damage, and thereby preventing tumor initiation and progression (Bose and Mukherjee, 2019).
Interventions targeting the gut microbiome, such as probiotics, prebiotics, synbiotics, FMT, and dietary modifications, have demonstrated considerable promise in preclinical models by reinforcing protective host barriers and mitigating oncogenic pathways. However, translating these compelling experimental findings into tangible clinical applications for cancer prevention remains a formidable challenge.
First, bridging the “translational valley” between preclinical models and human application is imperative. The striking efficacy of probiotics and synbiotics in rodent models of carcinogenesis (Table 1) has not been consistently mirrored in human trials. This disconnect likely stems from profound inter-species differences in gut microbiome composition and host immunity, as well as the staggering inter-individual variability of the human microbiome, which is shaped by genetics, long-term diet, and environmental exposures (Tsilimigras et al., 2017).
Second, there is an imperative to move beyond microbial correlation and establish causative mechanisms. Although numerous associations have been documented between specific bacteria (e.g., Bifidobacterium, Akkermansia) and improved anti-tumor immunity, the precise molecular messenger, whether the SCFAs like butyrate, unique metabolites like indole-3-lactic acid, or bacterial components themselves, often remain elusive (Smith et al., 2013; Zhang et al., 2023). Future research must, therefore, integrate multi-omics technologies (metagenomics, meta transcriptomics, metabolomics) with gnotobiotic models and sophisticated in vitro systems like organoids to dissect the precise mechanistic links between specific microbial strains, their metabolites, and host anti-tumor pathways (Sun et al., 2025). This mechanistic clarity is the basis for identifying reliable targets.
Third, the identification of key microbial molecules and pathways naturally paves the way for precision prevention. Mechanistic insights can be translated into clinical practice through diagnostics that screen for these specific microbial genes or metabolite profiles as biomarkers of increased cancer risk, enabling early detection of high-risk individuals. Personalized prevention strategies could include FMT from optimally screened donors to restore microbial balance or the development of engineered microbial consortia and “postbiotic” formulations, defined microbial metabolites that bypass the challenges of live bacterial delivery while directly exerting protective effect (Chen et al., 2019). Achieving this vision will require concerted, consortium-level efforts to standardize microbiome research methodologies, from sample collection to bioinformatics analysis, to ensure reproducibility, data comparability and accelerated discovery.
Finally, the scope of the gut microbiome’s influence demands a systemic perspective. Microbial metabolites are not confined to the intestinal lumen but circulate throughout the body, influencing distant organs. The gut-lung axis, for instance, where gut microbiota-derived SCFAs modulate immune responses in the lung, illustrates the potential for gut-focused interventions to prevent extraintestinal malignancies (Hanus et al., 2021). Similarly, microbiota-modified bile acids regulate hepatic NKT cell accumulation, shaping immune surveillance in HCC (Ma et al., 2018). These findings reveal a paradigm shift: modulating the gut microbiome is not merely a strategy for CRC prevention but a gateway to influencing systemic oncogenic risk and therapeutic efficacy across cancer types.
In summary, while our understanding of the gut microbiota’s role in cancer prevention is still evolving, the path forward is clear. By harnessing emerging technologies, fostering interdisciplinary collaboration, and prioritizing human trials, we can begin to strategically manipulate this intricate internal ecosystem. The ultimate goal is to integrate microbiome modulation into the framework of precision medicine, paving the way for novel, effective, and personalized strategies to reduce the global burden of cancer.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Albillos A. de Gottardi A. Rescigno M. (2020). The gut-liver axis in liver disease: pathophysiological basis for therapy. J. Hepatol. 72 (3), 558–577. 10.1016/j.jhep.2019.10.003 31622696 · doi ↗ · pubmed ↗
- 2Ali M. S. Hussein R. M. Gaber Y. Hammam O. A. Kandeil M. A. (2019). Modulation of JNK-1/beta-catenin signaling by Lactobacillus casei, inulin and their combination in 1,2-dimethylhydrazine-induced colon cancer in mice. RSC Adv. 9 (50), 29368–29383. 10.1039/c 9ra 04388 h 35528422 PMC 9071812 · doi ↗ · pubmed ↗
- 3Allen J. M. Mailing L. J. Niemiro G. M. Moore R. Cook M. D. White B. A. (2018). Exercise alters gut microbiota composition and function in lean and Obese humans. Med. Sci. Sports Exerc 50 (4), 747–757. 10.1249/MSS.0000000000001495 29166320 · doi ↗ · pubmed ↗
- 4Amin M. Navidifar T. Saeb S. Barzegari E. Jamalan M. (2023). Tumor-targeted induction of intrinsic apoptosis in colon cancer cells by Lactobacillus plantarum and Lactobacillus rhamnosus strains. Mol. Biol. Rep. 50 (6), 5345–5354. 10.1007/s 11033-023-08445-x 37155013 · doi ↗ · pubmed ↗
- 5Anderson J. L. Edney R. J. Whelan K. (2012). Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment. Pharmacol. Ther. 36 (6), 503–516. 10.1111/j.1365-2036.2012.05220.x 22827693 · doi ↗ · pubmed ↗
- 6Appleyard C. B. Cruz M. L. Isidro A. A. Arthur J. C. Jobin C. De Simone C. (2011). Pretreatment with the probiotic VSL#3 delays transition from inflammation to dysplasia in a rat model of colitis-associated cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 301 (6), G 1004–G 1013. 10.1152/ajpgi.00167.2011 21903764 PMC 3233787 · doi ↗ · pubmed ↗
- 7Arnone A. A. Cook K. L. (2022). Gut and breast microbiota as endocrine regulators of hormone receptor-positive breast cancer risk and therapy response. Endocrinology 164 (1), bqac 177. 10.1210/endocr/bqac 177 36282876 PMC 9923803 · doi ↗ · pubmed ↗
- 8Arpaia N. Campbell C. Fan X. Dikiy S. van der Veeken J. de Roos P. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504 (7480), 451–455. 10.1038/nature 12726 24226773 PMC 3869884 · doi ↗ · pubmed ↗