Gitelman Syndrome Presenting With Syncope and Treatment‐Refractory Hypokalemia in A Young Woman: A Case Report
Iyassu S. Melkie, Abenezer A. Wolde, Lulit Y. Mengesha, Rahwa A. Kinfe, Chernet T. Mengistie, Zenebwork Y. Gubai

TL;DR
A young woman with Gitelman syndrome experienced severe hypokalemia and syncope, showing the challenges in treating this rare genetic disorder.
Contribution
Highlights the diagnostic importance of Gitelman syndrome in young adults with treatment-resistant hypokalemia and metabolic alkalosis.
Findings
Patient showed severe hypokalemia, metabolic alkalosis, and hypomagnesemia consistent with Gitelman syndrome.
Despite potassium and magnesium supplementation, biochemical correction remained incomplete.
Genetic testing confirmed a pathogenic SLC12A3 variant, confirming the diagnosis.
Abstract
Gitelman syndrome (GS) is a rare autosomal recessive tubulopathy characterized by hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria. A 27‐year‐old woman presented with a witnessed syncopal episode, progressive weakness, and nausea. She reported a 3‐year history of muscle cramps, paresthesias, salt craving, and nocturia, with only transient correction of hypokalemia despite supplementation. Examination showed orthostatic hypotension and proximal muscle weakness, and ECG revealed flattened T and prominent U waves. Laboratory tests demonstrated severe hypokalemia (2.7 mmol/L), metabolic alkalosis, hypomagnesemia, renal potassium wasting, hypocalciuria, elevated renin and aldosterone, and a negative diuretic screen, consistent with GS. Severe hypokalemia is arrhythmogenic; ECG changes and syncope in this patient prompted monitored cardiac care and urgent correction. She was…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
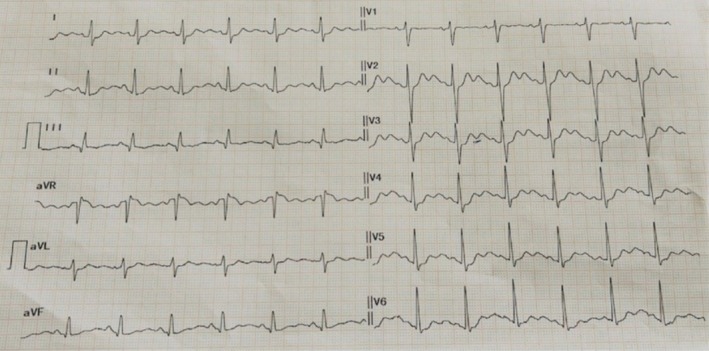 FIGURE 1
FIGURE 1| Laboratory/investigation | At admission | After 24 h | At discharge (day 5) | 8‐week follow‐up | Reference range |
|---|---|---|---|---|---|
| Hemoglobin | 13.2 g/dL | — | 13.1 g/dL | 13.0 g/dL | 12.0–16.0 g/dL |
| White blood cells | 6.5 × 109/L | — | 6.3 × 109/L | 6.4 × 109/L | 4.0–11.0 × 109/L |
| Platelets | 250 × 109/L | — | 248 × 109/L | 255 × 109/L | 150–400 × 109/L |
| MCV | 88 fL | — | 87 fL | 88 fL | 80–100 fL |
| Sodium (Na+) | 137 mmol/L | 136 mmol/L | 137 mmol/L | 137 mmol/L | 135–145 mmol/L |
| Potassium (K+) | 2.7 mmol/L | 3.2 mmol/L | 3.4 mmol/L | 3.3–3.7 mmol/L | 3.5–5.0 mmol/L |
| Chloride (Cl−) | 96 mmol/L | 98 mmol/L | 97 mmol/L | 98 mmol/L | 98–107 mmol/L |
| Bicarbonate (HCO3 −) | 33 mmol/L | 30 mmol/L | 28 mmol/L | 26–28 mmol/L | 22–28 mmol/L |
| Creatinine | 0.7 mg/dL | 0.7 mg/dL | 0.7 mg/dL | 0.7 mg/dL | 0.6–1.1 mg/dL |
| Magnesium (Mg2+) | 0.55 mmol/L | 0.75 mmol/L | 0.85 mmol/L | 0.90 mmol/L | 0.70–1.10 mmol/L |
| Calcium (total, corrected) | 9.1 mg/dL | 9.0 mg/dL | 9.0 mg/dL | 9.1 mg/dL | 8.5–10.2 mg/dL |
| Phosphate | 1.0 mmol/L | — | 1.1 mmol/L | 1.0 mmol/L | 0.8–1.5 mmol/L |
| AST | 22 U/L | — | 20 U/L | 21 U/L | 0–40 U/L |
| ALT | 18 U/L | — | 17 U/L | 18 U/L | 0–40 U/L |
| ALP | 68 U/L | — | 65 U/L | 66 U/L | 40–120 U/L |
| Total bilirubin | 0.7 mg/dL | — | 0.7 mg/dL | 0.7 mg/dL | 0.1–1.2 mg/dL |
| TSH | 1.8 mIU/L | — | 1.7 mIU/L | 1.8 mIU/L | 0.4–4.0 mIU/L |
| Free T4 | 1.1 ng/dL | — | 1.1 ng/dL | 1.1 ng/dL | 0.8–1.8 ng/dL |
| INR | 1.0 | — | 1.0 | 1.0 | 0.9–1.2 |
| aPTT | 32 s | — | 31 s | 32 s | 25–40 s |
| Arterial blood gas (room air) | pH 7.48, pCO2 41 mmHg, HCO3 − 33 mmol/L | pH 7.44, pCO2 40 mmHg | pH 7.40, HCO3 − 28 mmol/L | pH 7.38, HCO3 − 26 mmol/L | pH 7.35–7.45 |
| Urinalysis | Specific gravity 1.010; no protein; no blood; no glucose | — | — | — | — |
| 24‐h urinary potassium | 65 mmol/24 h | — | — | — | Elevated in renal wasting |
| Spot urine Ca/Cr ratio | 0.04 (low) | — | — | — | Low in hypocalciuria |
| Plasma renin activity | 6.5 ng/mL/h | — | — | — | 0.2–2.8 ng/mL/h |
| Plasma aldosterone | 37 ng/dL | — | — | — | 4–31 ng/dL |
| Urine diuretic screen | Negative (performed during admission) | — | — | — | — |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIon Transport and Channel Regulation · Ion channel regulation and function · Cardiac electrophysiology and arrhythmias
Introduction
1
Gitelman syndrome (GS) is an autosomal recessive salt‐wasting renal tubulopathy characterized biochemically by hypokalemic metabolic alkalosis, hypomagnesemia, hypocalciuria, and secondary hyperaldosteronism [1]. It is rare, with an estimated prevalence of ~1 in 40,000 individuals [2]. GS is caused by bi‐allelic loss‐of‐function mutations in the SLC12A3 gene on chromosome 16q13, which encodes the thiazide‐sensitive Na^+–Cl^– cotransporter (NCC) in the distal convoluted tubule [3, 4]. Inactivating NCC mutations impair renal NaCl reabsorption, leading to mild volume depletion, chronic stimulation of the renin–angiotensin–aldosterone system, and renal K^+ and Mg^2+ wasting [1, 5].
Patients with GS typically present in late childhood, adolescence, or early adulthood [6]. Clinical manifestations are variable. Many patients are asymptomatic or only mildly symptomatic, often being diagnosed incidentally on routine blood tests [4, 6]. When present, symptoms reflect electrolyte depletion and volume contraction, including salt craving, fatigue, muscle weakness or cramps, paresthesias, tetany, and polyuria/nocturia [2, 4, 5]. Patients may have low or normal blood pressure due to chronic salt wasting [7]. Importantly, the chronic hypokalemia of GS frequently prolongs ventricular repolarization. While marked arrhythmias are uncommon [8], QT prolongation is often seen, and rare cases of syncope or sudden cardiac arrest have been reported [2, 8].
Diagnosis of GS relies on characteristic biochemical findings together with exclusion of other causes of hypokalemic alkalosis. Key laboratory features include persistent hypokalemia, metabolic alkalosis, hypomagnesemia, and low urinary calcium excretion [1, 4]. Plasma renin and aldosterone are typically elevated in the setting of normotension [7]. A formal urine diuretic screen and urinary electrolyte indices help exclude surreptitious diuretic use [9]. Genetic testing of SLC12A3 can confirm the diagnosis in most cases [1, 5].
Management of GS centers on liberalizing salt intake and aggressive electrolyte supplementation [9, 10]. Lifelong high‐dose oral potassium and magnesium are required, often in combination with potassium‐sparing diuretics such as amiloride or spironolactone to reduce renal K^+ losses [11]. Patients are encouraged to eat a high‐sodium, high‐potassium, and high‐magnesium diet [10]. Careful cardiac monitoring is recommended when hypokalemia is severe [12]. With treatment, many patients achieve near‐normal electrolytes; however, partial hypokalemia often persists due to renal losses [4, 9, 11].
We report a case of GS in a young woman who presented with syncope and severely refractory hypokalemia to highlight the clinical features, diagnostic considerations, and treatment challenges of this disorder.
Clinical History/Examination
2
A 27‐year‐old Ethiopian woman presented to the emergency department after a witnessed syncopal episode and 48 h of progressive generalized weakness, nausea, and two episodes of near‐syncope. She reported a three‐year history of intermittent muscle cramps, distal paresthesia, longstanding salt craving, polyuria, and nocturia (typically awakening twice nightly), and multiple prior outpatient courses of oral potassium replacement (40–80 mmol/day) that produced only transient correction. She denied vomiting, diarrheal illness, laxative use, prescribed or over‐the‐counter diuretics, herbal remedies, or illicit drug use. Her past medical history was otherwise unremarkable. Family history was notable for a maternal uncle described as having “similar cramps” treated with potassium tablets but without a formal diagnosis; there was no known consanguinity.
On examination, she was thin (BMI 19.8 kg/m^2^), alert and oriented, afebrile, pulse 88/min, and blood pressure 100/68 mmHg supine with a 12 mmHg orthostatic fall in systolic pressure on standing and a mild compensatory tachycardia. Cardiorespiratory and abdominal examinations were unremarkable. Neurological examination demonstrated proximal greater than distal weakness (hip flexion and shoulder abduction 4/5 bilaterally; distal strength 4+/5), intact sensation, and normal deep tendon reflexes; gait was mildly unsteady on heel walking, attributable to weakness. Admission 12‐lead ECG showed sinus rhythm ~90/min with flattened T waves, prominent U waves, and a borderline prolonged QTc (Figure 1).
Electrocardiogram on admission showing sinus rhythm with flattened T waves, prominent U waves, and borderline QT prolongation, consistent with hypokalemia.
Differential Diagnosis, Investigations, and Treatment
3
Initial laboratory testing confirmed marked hypokalemia (serum K^+^ 2.7 mmol/L) with metabolic alkalosis (serum HCO_3_ ^−^ 33 mmol/L), hypomagnesemia (serum Mg^2+^ 0.55 mmol/L), normal serum sodium, and preserved renal function (serum creatinine 0.7 mg/dL). Spot urine testing and a timed collection demonstrated renal potassium wasting (24‐h urinary K^+^ 65 mmol/24 h) and hypocalciuria (spot urine Ca/Cr ratio 0.04). Plasma renin activity (6.5 ng/mL/h) and aldosterone (37 ng/dL) were both elevated in the context of normal/low blood pressure, consistent with a secondary hyperreninemic state. A formal urine diuretic screen was performed during admission and returned negative (Table 1), supporting endogenous renal salt‐wasting rather than surreptitious diuretic use. Thyroid function and random cortisol were within reference ranges. Based on the combination of hypokalemia, metabolic alkalosis, hypomagnesemia, hypocalciuria, elevated renin and aldosterone, normal blood pressure, and a negative diuretic screen, a clinical diagnosis of Gitelman syndrome was made.
Because of symptomatic and ECG‐documented severe hypokalemia, she was admitted to a monitored bed and received continuous cardiac monitoring. Acute management included intravenous potassium chloride administered in divided doses (total 120 mmol over 24 h) and intravenous magnesium sulfate 2 g over 4 h with serial electrolyte monitoring; serum potassium rose to 3.2 mmol/L and magnesium to 0.75 mmol/L with partial symptomatic improvement. For ongoing management, she was commenced on oral magnesium oxide 400 mg three times daily, sustained‐release potassium chloride 40 mmol twice daily, and amiloride (initiated 5 mg daily and titrated to 10 mg daily) to reduce renal potassium wasting. She received counseling to moderately liberalize dietary sodium, increase potassium‐ and magnesium‐rich foods, and avoid substances known to exacerbate kaliuresis (excessive caffeine, licorice). No adverse events occurred during replacement, and renal function remained stable. The ECG abnormalities resolved, and the patient reported partial symptomatic improvement; therefore, no additional potassium‐sparing agent (e.g., spironolactone) or NSAIDs were initiated at discharge.
Outcome and Follow‐Up
4
The patient was discharged on hospital day five with serum potassium 3.4 mmol/L and magnesium 0.85 mmol/L and arranged for close outpatient follow‐up. Over eight weeks of outpatient care, adherence to oral supplements and amiloride produced notable improvement in cramps, fatigue, and presyncopal symptoms, with no further syncopal episodes, allowing return to usual activities. However, serum potassium remained low‐normal to borderline (3.3–3.7 mmol/L), consistent with the chronic biochemical pattern of Gitelman syndrome.
Genetic testing at follow‐up was performed using a targeted NGS tubulopathy panel (mean coverage ~150×, > 98% of targets ≥ 20×). Two compound heterozygous pathogenic variants were identified in SLC12A3: c.179C> T (p.Thr60Met) in exon 1, and c.2221G> A (p.Gly741Arg) in exon 18. Both variants were confirmed by Sanger sequencing. The laboratory classified each as pathogenic according to ACMG criteria (e.g., PM1, PM2, PP3). Both variants are rare in gnomAD (allele frequencies ~0.0001–0.0003) and are reported in ClinVar. No copy‐number alterations were detected. Parental segregation testing was not performed. This confirmed the diagnosis of Gitelman syndrome. Limitations of the inpatient workup were therefore addressed: the negative urine diuretic screen obtained during admission helped exclude surreptitious diuretic use, and genetic confirmation was obtained subsequently through the outsourced test.
Discussion
5
This patient's presentation illustrates the key features and management challenges of GS. Persistent refractory hypokalemia is a hallmark of GS. In fact, chronic hypokalemia is “the main finding” in affected patients [6]. Despite aggressive replacement, most GS patients remain mildly hypokalemic, requiring ongoing supplementation. In the long term, GS is a lifelong condition: serum K^+ and Mg^2+ typically improve with treatment but rarely normalize completely [6, 9]. There are limited prospective data on the longitudinal course, but existing reports indicate that GS does not resolve with age, and symptom burden (fatigue, cramps) often persists into adulthood [4, 13].
Differentiating GS from other causes of hypokalemic alkalosis is critical. In this case, exclusion of Bartter syndrome and diuretic abuse was important. Compared with GS, classic Bartter syndromes present much earlier (often antenatally or in infancy) and commonly feature hypercalciuria [5, 6, 10, 14]. In contrast, GS patients typically present later with hypocalciuria and normal/low blood pressure [14]. Measurement of 24‐h urinary calcium can help distinguish GS (low urinary Ca) from Bartter (high urinary Ca) [5, 14]. Surreptitious diuretic use must also be considered in unexplained hypokalemia. A comprehensive diuretic screen and the context of chronic symptoms help exclude this; in our patient, a negative diuretic screen supported a renal tubular etiology [9].
Therapeutically, GS often requires a multi‐pronged approach. The cornerstone is high‐dose electrolyte supplementation. Oral potassium chloride and magnesium oxide were used in our patient as recommended [9, 13]. Adjunctive therapies aim to reduce renal potassium wasting. Potassium‐sparing diuretics such as amiloride or spironolactone counteract hyperaldosteronism and have been shown to improve serum K^+ in GS [10, 11]. Indeed, antialdosterone therapy (spironolactone and amiloride) has been reported to significantly raise serum K^+ and reduce urinary K^+ excretion in GS patients [11]. In one series, combined spironolactone/amiloride treatment raised mean serum K^+ from ~2.6 to 3.4 mmol/L (p < 0.001) and reduced renal K^+ clearance [15]. Amiloride and spironolactone are both reasonable; the choice depends on patient tolerance and side effect profiles [10, 15]. In our patient, amiloride was initiated together with oral magnesium oxide and sustained‐release potassium chloride after acute intravenous replacement; serum K rose to 3.2 mmol/L and Mg to 0.75 mmol/L, and the ECG abnormalities normalized. Because symptoms and ECG changes improved while on amiloride, we judged that escalation to an additional potassium‐sparing agent was not indicated at discharge.
Dietary counseling (including liberal salt intake, high‐potassium foods, and avoidance of licorice or excessive caffeine) is also advised [14, 16]. In refractory cases, adjunctive therapies (e.g., prostaglandin synthesis inhibitors) are sometimes considered, though their benefit is clearer in Bartter variants than in GS [11, 17]. COX inhibitors (most commonly indomethacin) can raise serum potassium in some series and may be considered for selected refractory Gitelman cases, but indomethacin was the most effective in small studies and is limited by gastrointestinal intolerance and reductions in eGFR; therefore, NSAIDs should be reserved for refractory patients after nephrology review and monitoring [11].
Regarding outcomes, GS is generally considered to have a benign long‐term prognosis [9]. Life expectancy is usually normal, and most patients avoid life‐threatening complications with treatment [4]. However, quality of life can be significantly affected by chronic symptoms. For example, a large GS cohort found fatigue and muscle cramps in over 80% of patients [13], and many report salt craving and nocturia. Even with treatment, GS patients often report ongoing tiredness and reduced well‐being, underscoring the chronicity of the condition [4, 9, 13]. Persistent hypokalemia can prolong the QT interval; although serious ventricular arrhythmias are uncommon, vigilance is warranted [8]. Indeed, isolated cases of syncope and sudden cardiac arrest have been attributed to untreated GS [2, 8]. GS has also been associated with metabolic perturbations: recent studies suggest carriers of SLC12A3 mutations may have altered glucose metabolism, and GS patients appear to have a higher incidence of type 2 diabetes compared with controls [18]. There are even reports of extrarenal features such as chondrocalcinosis in some GS patients, likely related to chronic hypomagnesemia [19]. Renal function is typically preserved, but rare cases of chronic kidney disease have been reported, possibly secondary to long‐term hypokalemia and hypovolemia [4, 10].
Gitelman syndrome shows marked genotype–phenotype heterogeneity: different SLC12A3 variants produce variable biochemical profiles (including magnesium levels), age at onset, and occasional extrarenal associations; precise genotyping therefore aids prognostication and comparison with reported cases [1, 20]. Detailed variant reporting (c./p. notation, exon and zygosity) enables nuanced genotype–phenotype comparisons, for example, Koca et al. documented an atypical presentation of GS with autoimmune thyroiditis [20].
Our therapeutic goals in this patient are sustained symptom control, prevention of arrhythmia, and maintenance of near‐normal potassium and magnesium levels. She will continue oral supplementation and amiloride with periodic electrolyte and ECG monitoring. Escalation to spironolactone or NSAIDs would be considered only if symptoms recur or biochemical control becomes inadequate. In summary, this case of GS highlights the importance of suspecting GS in young patients with unexplained hypokalemic metabolic alkalosis. Its management requires aggressive and multidisciplinary therapy. While GS often carries a favorable prognosis in terms of mortality, the burden of chronic electrolyte management and symptomatology is nontrivial. Ongoing follow‐up is essential to monitor electrolytes, growth (if pediatric), blood pressure, and ECG changes.
Conclusion
6
Gitelman syndrome is a lifelong salt‐wasting tubulopathy that often presents in young adults with muscle cramps, salt craving, and refractory hypokalemia. Our patient's syncope was precipitated by severe hypokalemia due to GS. This case reinforces that clinicians should suspect GS when unexplained hypokalemia is accompanied by metabolic alkalosis, hypomagnesemia, and hypocalciuria in a normotensive patient. Early recognition allows genetic confirmation and the institution of tailored therapy. Despite maximal treatment with potassium and magnesium supplements and potassium‐sparing agents, many GS patients continue to exhibit low‐normal serum potassium. Nonetheless, aggressive electrolyte management can alleviate symptoms, reduce arrhythmic risk, and improve quality of life in this rare disorder.
Author Contributions
Iyassu S. Melkie: conceptualization, visualization, writing – original draft. Abenezer A. Wolde: visualization, writing – review and editing. Lulit Y. Mengesha: data curation, resources. Rahwa A. Kinfe: data curation, resources. Chernet T. Mengistie: writing – original draft, writing – review and editing. Zenebwork Y. Gubai: supervision, writing – review and editing.
Funding
The authors has nothing to report.
Ethics Statement
IRB review and approval were waived for this case report.
Consent
Written informed consent was obtained from the patient for publication of the case details and accompanying images.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1N. Li and H. F. Gu , “Genetic and Biological Effects of SLC 12A 3, a Sodium‐Chloride Cotransporter, in Gitelman Syndrome and Diabetic Kidney Disease,” Frontiers in Genetics 13 (2022): 799224.35591852 10.3389/fgene.2022.799224 PMC 9111839 · doi ↗ · pubmed ↗
- 2A. Kondo , C. Nagano , S. Ishiko , et al., “Examination of the Predicted Prevalence of Gitelman Syndrome by Ethnicity Based on Genome Databases,” Scientific Reports 11, no. 1 (2021): 16099.34373523 10.1038/s 41598-021-95521-6PMC 8352941 · doi ↗ · pubmed ↗
- 3K. Nozu , T. Yamamura , T. Horinouchi , et al., “Inherited Salt‐Losing Tubulopathy: An Old Condition but a New Category of Tubulopathy,” Pediatrics International 62, no. 4 (2020): 428–437.31830341 10.1111/ped.14089 · doi ↗ · pubmed ↗
- 4J. Fujimura , K. Nozu , T. Yamamura , et al., “Clinical and Genetic Characteristics in Patients With Gitelman Syndrome,” Kidney Int Rep 4, no. 1 (2019): 119–125.30596175 10.1016/j.ekir.2018.09.015PMC 6308995 · doi ↗ · pubmed ↗
- 5K. P. Schlingmann and J. H. F. De Baaij , “The Genetic Spectrum of Gitelman(−Like) Syndromes,” Current Opinion in Nephrology and Hypertension 31, no. 5 (2022): 508–515.35894287 10.1097/MNH.0000000000000818 PMC 9415222 · doi ↗ · pubmed ↗
- 6D. Mamalis , T. Stratigou , N. Vallianou , G. Ioannidis , and T. Apostolou , “Persistent Hypokalemia due to a Rare Mutation in Gitelman's Syndrome,” Saudi Journal of Kidney Diseases and Transplantation 31, no. 1 (2020): 259.32129221 10.4103/1319-2442.279949 · doi ↗ · pubmed ↗
- 7W. Ji , J. N. Foo , B. J. O'Roak , et al., “Rare Independent Mutations in Renal Salt Handling Genes Contribute to Blood Pressure Variation,” Nature Genetics 40, no. 5 (2008): 592–599.18391953 10.1038/ng.118PMC 3766631 · doi ↗ · pubmed ↗
- 8A. Bettinelli , C. Tosetto , G. Colussi , G. Tommasini , A. Edefonti , and M. G. Bianchetti , “Electrocardiogram With Prolonged QT Interval in Gitelman Disease,” Kidney International 62, no. 2 (2002): 580–584.12110021 10.1046/j.1523-1755.2002.00467.x · doi ↗ · pubmed ↗