Mild Generation of Highly Nucleophilic N‐Heterocyclic Carbene Boryl Anion From Neutral sp2–sp3 Diboron Reagents and Its Applications in Nucleophilic Borylation
Weixuan Sun, Peiqi Zhang, Hairong Lyu

TL;DR
A highly reactive boryl anion is generated at room temperature and used to efficiently borylate a wide range of electrophiles.
Contribution
A new method for generating a highly nucleophilic NHC boryl anion at room temperature using a neutral diboron reagent.
Findings
The NHC boryl anion is generated in situ at room temperature using (NHC)BH2Bpin and KOtBu.
The anion shows broad reactivity with diverse electrophiles, including traditionally inert ones.
DFT calculations confirm the anion's nucleophilicity and reaction mechanisms.
Abstract
Due to its high activity, the synthesis or generation of the N‐heterocyclic carbene (NHC) boryl anion remains challenging, with only limited methods available. Traditional stabilization through bulky ligands or aromatic systems restricts its reactivity as a boron synthon. In this work, we present a straightforward strategy for the in situ generation of an NHC boryl anion at room temperature through the reaction of neutral sp2–sp3 diboron reagent (NHC)BH2Bpin with KO t Bu. This less sterically hindered and strongly nucleophilic boryl anion demonstrates broad reactivity with diverse electrophiles, including those that are unreactive with other types of boryl anions or traditional diboron reagents. These nucleophilic borylation reactions enable the synthesis of a diverse array of four‐coordinate organoboron compounds. Detailed mechanistic studies and DFT calculations confirm the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 SCHEME 1
SCHEME 1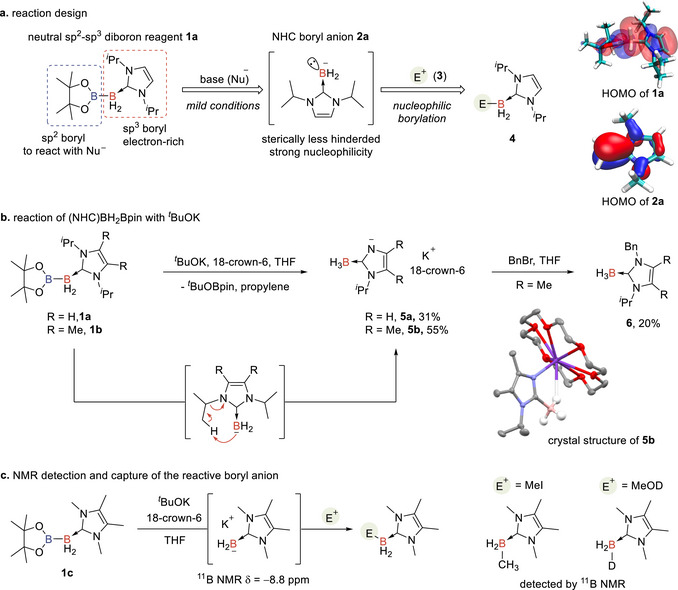 SCHEME 2
SCHEME 2 SCHEME 3
SCHEME 3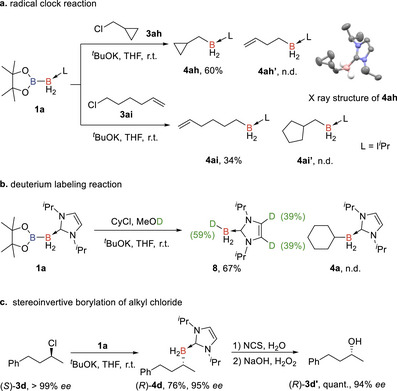 SCHEME 4
SCHEME 4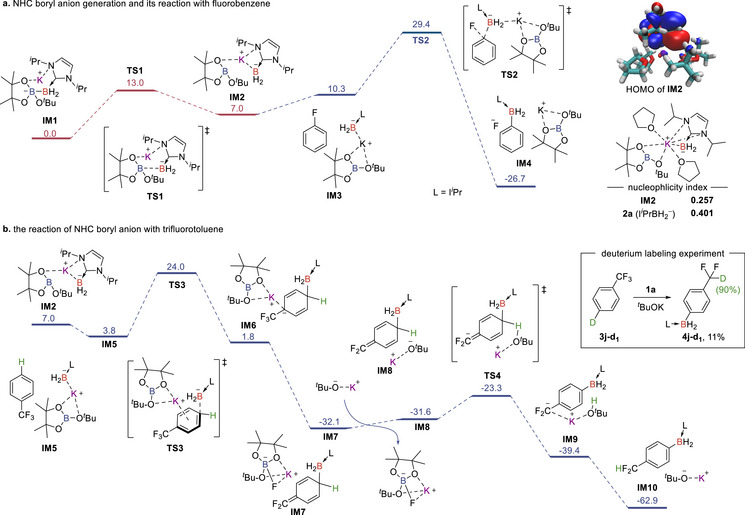 FIGURE 1
FIGURE 1|
|
|
|
- —Chinese University of Hong Kong10.13039/501100004853
- —Research Grants Council of HKSAR
- —State Key Laboratory of Synthetic Chemistry
- —Croucher Foundation10.13039/501100001692
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganoboron and organosilicon chemistry · Catalytic Cross-Coupling Reactions · N-Heterocyclic Carbenes in Organic and Inorganic Chemistry
Introduction
1
Since E. J. Corey introduced the concept of synthons as retrosynthetic building blocks in 1967 [1, 2], expectations for these reagents have grown well beyond their basic role. Ideally, a modern synthon is easy to prepare from affordable starting materials, avoids highly toxic or hazardous reagents, can be handled without special precautions, and shows good compatibility to enable flexible synthesis. Identifying synthons that satisfy these criteria remains an active area of interest. The same considerations apply in boron chemistry: access to reactive yet user‐friendly boron synthons would further broaden the scope and utility of organoboron compounds.
N‐heterocyclic carbene stabilized borane (NHC)BH_3_ is relatively electron‐rich and smoothly undergoes dehydridation with acids to form borenium species (NHC)BH_2_ ^+^ (Scheme 1a), which serves as an electrophilic boryl intermediate in borylation reactions [3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. Moreover, (NHC)BH_3_ can be subjected to single electron oxidation or hydrogen atom transfer to give a three‐coordinated boryl radical, which often displays nucleophilic properties and is widely used in borylation of electron‐deficient unsaturated substrates via a radical addition process (Scheme 1b) [16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27]. These developments provide fundamental insights and useful sp^3^ boryl synthons for the preparation of four‐coordinate organoboron compounds.
Representative N‐heterocyclic carbene stabilized sp3 boryl synthons and their applications in borylation reactions and four‐coordinate organoboron compound synthesis.
In sharp contrast, the exploration of NHC boryl anion species remains significantly underdeveloped (Scheme 1c). Due to the weak electronegativity of boron (∼2.0), it has traditionally presented significant challenges in the formation of its nucleophilic boryl anions [28, 29, 30, 31, 32, 33, 34, 35, 36]. The initial success in stabilizing and isolation of these highly active species was marked by Yamashita and Nozaki's pioneering synthesis of boryl lithium compounds in 2006 [37]. Subsequent efforts have explored various stabilization strategies, including the use of bulky ligands [37, 38, 39, 40, 41, 42, 43, 44], incorporation into aromatic systems [37, 41, 43, 44, 45, 46, 47, 48, 49], and the addition of electron‐withdrawing groups [38, 50, 51, 52, 53]. These methods, while effective in stabilizing boryl anions, may inadvertently compromise their nucleophilic capability. In 2010, Braunschweig and coworkers first isolated and characterized a SIMes (SIMes = 1,3‐bis(2,4,6‐trimethylphenyl)‐imidazolin‐2‐ylidene) stabilized borole monoanion through the reduction of corresponding boryl chloride with KC_8_, which exhibited reactivity with electrophile iodomethane [46]. In the same year, Lacôte and coworkers reported an approach for the generation of a boryl anion protected with the bulky IDip (IDip = 1,3‐bis(2,6‐diisopropylphenyl)‐imidazole‐2‐ylidene) via the reduction of boryl iodide (IDip)BH_2_I with lithium di‐tert‐butylbiphenylide (LDBB) at −78°C [54]. This (IDip)BH_2_‾ could not be isolated, and its nucleophilic reactivities were demonstrated via the in situ reactions with organic halides and electron‐deficient aromatics.
In this context, the use of a highly nucleophilic NHC boryl anion as a boron synthon is anticipated to enable new borylation reactions and expand product diversity in the synthesis of organoboron compounds. To achieve this, a new strategy and robust boron precursors are greatly needed, which should 1) avoid the use of harsh conditions, such as strong reducing agents and cryogenic temperatures, and 2) exhibit remarkable nucleophilic properties, allowing reactions with a wide range of electrophiles, including those that were previously challenging to access.
Results and Discussion
2
Generation of Boryl Anions
2.1
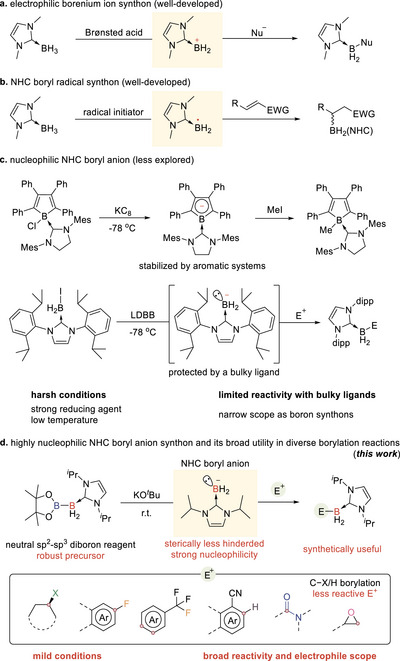
Recently, we reported a neutral sp^2^–sp^3^ diborane (I* ^i^ Pr)BH_2_Bpin (I ^i^ *Pr = 1,3‐diisopropylimidazol‐2‐ylidene), which exhibits good stability toward air and moisture and can be used as a borylation reagent in organic synthesis [55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69]. In our prior studies, this reagent effectively engaged in copper‐catalyzed borylation of unsaturated substrates through the intermediacy of a key Cu−B(sp^3^) complex [55, 57]. Here, we envision that if the sp^2^–sp^3^ diborane can serve as a robust precursor for the generation of a strongly nucleophilic NHC boryl anion synthon under transition metal‐free conditions to unlock new borylation reactions (Scheme 1d).
Considering the unique structure and electronic properties of this sp^2^–sp^3^ diboron reagent, specifically, the sp^3^ boryl is stabilized by a less‐bulky, strong electron‐donating I* ^i^ Pr ligand and is electron‐rich, while the sp^2^ boryl group is electron‐deficient and electrophilic, we propose that the sp^2^ boryl can be coordinated by a base, triggering the heterolytic cleavage of the B−B bond and leading to the in situ generation of an I ^i^ *Pr‐stabilized boryl anion (Scheme 2a). Compared to the reduction of NHC boryl halides with strong reductants of LDBB or KC_8_, this strategy is anticipated to provide a milder approach for generating NHC boryl nucleophiles. Beyond the strong nucleophilicity, the low steric hindrance of the boryl anion may further expand its reactivity toward relatively inert or less electron‐deficient substrates, potentially expanding their applications as boron synthons in organic synthesis (Scheme 1d). In our initial trial, * ^t^ *BuOK was used as the base to facilitate B─B bond heterolytic cleavage, thereby generating the boryl anion through the elimination of * ^t^ *BuOBpin. In the reaction of diborane 1a or 1b with * ^t^ *BuOK in the presence of 18‐crown‐6, colorless crystals 5a and 5b were obtained. The structure of compound 5b was further confirmed via X‐ray analysis (Scheme 2b) [70]. It is deduced that the NHC boryl anion was initially formed. However, due to the strong basicity, it rapidly underwent an intramolecular pericyclic reaction, affording the stable potassium amide 5b, and releasing propylene as a by‐product. Compound 5b was subsequently reacted with the electrophile benzyl bromide to yield compound 6 [28]. These results indicate that the neutral sp^2^–sp^3^ diborane (NHC)BH_2_Bpin might serve as a suitable precursor for generating the NHC boryl anion under basic conditions.
Generation of NHC boryl anion from the neutral sp2–sp3 diboron reagent (NHC)BH2Bpin.
To detect the possible NHC boryl anion by ^11^B NMR, diboron reagent 1c, which lacks β‐hydrogens for the pericyclic reaction, was reacted with * ^t^ *BuOK. A predominant triplet signal at δ = −8.8 ppm appeared in the ^11^B NMR spectrum. Comparison with reported chemical shifts for carbene‐stabilized dihydroboryl anions (^11^B NMR δ = −4.7 ppm, δ = −18.1 ppm) [39, 54] suggests that this signal likely corresponds to a similar species. Upon subsequent treatment with electrophiles such as MeI and MeOD, this triplet was completely converted into signals corresponding to methyl borane and deuterated borane, respectively (Scheme 2c). These results provide evidence for the generation of a reactive boron species that acts as both a strong base and a nucleophile, which is presumably identified as the NHC boryl anion (see Supporting Information, Section 6 and Figure S2 for details).
Nucleophilic Borylation
2.2
In this connection, nucleophilic borylation of various electrophiles (E^+^) was subsequently evaluated using (I* ^i^ *Pr)BH_2_Bpin 1a as a robust precursor of the NHC boryl anion synthon (Table 1). In the presence of * ^t^ *BuOK, 1a underwent a nucleophilic substitution reaction with alkyl chlorides, affording alkyl boranes in satisfactory yields (4a‐4e). The S_N_Ar reaction of 1a with phenyl fluoride and substituted aryl fluorides also went well, producing aryl NHC boranes (4f‐4i) in moderate to good yields. The comparable reactivity with aryl fluorides containing electron‐donating groups indicates the strong nucleophilicity of the in situ generated NHC boryl anion.
TABLE 1: Nucleophilic borylation using (I i Pr)BH2Bpin (1a) as the NHC boryl anion precursor.
Additionally, reactions of 1a with trifluoromethyl arenes were explored. Trifluorotoluene, 4‐(trifluoromethyl)pyridine and 3‐(trifluoromethyl)pyridine reacted well, yielding defluoroborylation products 4j‐4l in 55% to 66% yields and good regioselectivity. Subsequent oxidation of 4l using H_2_O_2_ provided 4‐(difluoromethyl)‐2(1H)‐pyridinone 7 in 70% yield. It is noteworthy that diboron reagent 1a was also applied to achieve the direct C3 borylation of cyanoarenes [54] under the metal‐free conditions (4m‐4o). Moreover, the in situ generated boryl anion demonstrated nucleophilic reactivity with N‐methyl‐N‐phenylpivalamide, efficiently producing acyl borane 4p. Additionally, the nucleophilic attack of the boryl anion on an epoxide resulted in the ring‐opening product 4q with a yield of 40%.
The broad reactivity of the (I* ^i^ Pr)BH_2_Bpin (1a) is likely attributed to the strong nucleophilicity and low steric hindrance of the (I ^i^ *Pr)BH_2_‾ intermediate. Control experiments demonstrated that, under the same reaction conditions, B_2_pin_2_ did not react with alkyl chloride or trifluoromethyl pyridine (Scheme 3a) [71, 72]. Additionally, the in situ generated, more sterically bulky IDipBH_2_Li showed no reactivity with aryl fluoride and trifluoromethyl pyridine (Scheme 3b, see Supporting Information Figure S1 for details).
Control experiments using other nucleophilic boryl precursor and boron reagent.
To further expand the scope of electrophiles to include more inert ones, particularly aryl fluorides and trifluoromethyl arenes, which are unreactive toward other types of boryl anions, we employed 1c as the boryl anion precursor (Table 2). The in situ generated boryl anion derived from 1c is proposed to exhibit higher reactivity due to the less sterically hindered IMe_4_ (IMe_4_ = 1,3‐dihydro‐1,3,4,5‐tetramethyl‐2H‐imidazol‐2‐ylidene) ligand. S_N_Ar reactions of 1c with p‐fluorotoluene and p‐fluoroanisole afforded the corresponding aryl NHC boranes in moderate yields (4r, 4s). Notably, the more sterically hindered 2‐fluorobiphenyl and 3‐fluorobiphenyl afforded the desired products 4t and 4u in 62% and 66% yields, respectively. Additionally, a thiophenyl‐substituted aryl fluoride was also compatible, yielding 4v in 40% yield. The use of 1c further expanded the scope of trifluoromethyl arenes. Substrates substituted with both electron‐withdrawing (−F) and electron‐donating (−OMe) groups selectively yielded para‐defluoroborylation products (4w, 4x). Additionally, trifluoromethyl benzenes containing heterocyclic groups, such as benzofuran and thiophene, reacted with 1c to generate difluoromethyl aryl borane derivatives with moderate yields (4y, 4z). Moreover, the trifluoromethyl arene derived from a natural product (L)‐citronellol furnished the corresponding product 4aa.
In the one‐pot reactions, some reactive electrophiles may directly react with * ^t^ *BuOK, resulting in low yields or trace amounts of the desired borylation products. To address this, diboron 1c was first treated with a base in the presence of 18‐crown‐6 to generate the boryl anion, which was subsequently reacted with electrophiles. Using this protocol, various alkyl bromides reacted to afford alkyl boranes (4ac‐4ae). Diethyl carbonate selectively yielded the mono‐substituted acyl borane 4ab with satisfactory efficiency, indicating efficient conversion of 1c to the boryl anion in the first step. Additionally, treatment with TMSCl successfully generated the corresponding silylborane 4af, while the reaction of 1c with an aldehyde yielded the α‐hydroxyl borane product 4ag, albeit with a lower yield of 16%.
Mechanistic Study and DFT Calculation
2.3
To gain deeper insight into the reaction mechanism and further verify the involvement of boryl anion in the reaction pathway, we first performed the mechanistic study of the S_N_2 borylation reaction. A radical clock experiment was conducted using (chloromethyl)cyclopropane (3ah) as the electrophile [73, 74, 75, 76]. The alkyl borane 4ah was obtained as the sole product, while the ring‐opening product 4ah’ was not detected. Similarly, the reaction with 6‐chloro‐1‐hexene did not afford the cyclized product 4ai’. These results suggest that a radical pathway might not be involved in the reaction process (Scheme 4a).
Mechanistic study of the SN2 borylation.
We then examined the generation of the basic NHC boryl anion via a quenching experiment. The reaction mixture of 1a, KO* ^t^ *Bu, and cyclohexyl chloride was immediately quenched with MeOD before the S_N_2 reaction started. Instead of the detection of 4a, deuterated NHC borane 8 was obtained in 67% yield, with a deuteration rate of 59% on boron. A 39% deuteration rate was also observed on the NHC, which indicates a possible deprotonation of the C−H on NHC by the boryl anion. Since the hydrogen atom transfer (HAT) from MeOD by a boron radical species is not feasible, this result suggests that the reaction is likely an acid–base reaction involving a basic boryl anion (Scheme 4b).
More importantly, when the enantiopure alkyl chloride (S)‐3d (> 99% ee) was reacted with diboron 1a under standard conditions, the stereoinvertive borylation product (R)‐4d was obtained in 76% yield with 95% ee. The configuration of (R)‐4d was confirmed by in situ oxidation, resulting the alcohol (R)‐3d’ (Scheme 4c) with 94% ee. This result provides solid evidence for the S_N_2‐type mechanism in the nucleophilic borylation of alkyl chlorides and further supports the generation of an NHC‐boryl anion in the reaction pathway. It also represents the first example of transition‐metal‐free, stereospecific nucleophilic borylation of unactivated alkyl halides [77, 78, 79, 80], potentially offering a straightforward method for the synthesis of chiral alkyl boranes.
DFT computation was carried out to further understand the reaction mechanism (Figure 1), especially for the unique reactivities exhibited by the boryl anion 2a toward aryl fluorides and trifluoromethyl arenes, which have not been observed and explored with other types of boryl anions [37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54].
Mechanistic investigation and DFT‐calculated pathways for NHC boryl anion generation and its reactions with phenyl fluoride and trifluorotoluene. DFT computation on M06‐L / 6–31G(d,p) level. Explicit THF molecules around the potassium ion are omitted for clarity. Relative free energies are given in kcal/mol.
The DFT calculations were initiated with the borate intermediate IM1, formed from the reaction of KO* ^t^ *Bu and diborane 1a. The calculated energy barrier for B─B bond heterolytic cleavage was relatively low (13.0 kcal/mol), leading to the formation of the boryl potassium complex IM2. The highest occupied molecular orbital (HOMO) of IM2 was primarily localized on the p orbital of the boron atom bonded to the NHC, indicating a significant negative charge on the boron atom. Furthermore, natural fragment bonding orbital (NFBO) [81] analysis revealed a high electron distribution of 0.92 on boron, demonstrating its superior anionic character (see Figure S3 for details). The calculated nucleophilicity index [82] of IM2 (0.257) and the anionic fragment 2a (0.401) are also higher than those of other reported boryl anion complexes [37, 39, 51, 54] (see Figure S4 for details). These computational findings align well with the strong nucleophilicity and special reactivities of the boron anion observed experimentally.
When boron anion IM2 reacted with phenyl fluoride, it readily attacked phenyl fluoride via an S_N_Ar‐type mechanism to produce the arylborane product. A single transition state was identified, with no intermediate observed, as verified by an intrinsic reaction coordinate (IRC) scan (Figure 1a).
For the reactions with different trifluoromethyl arenes, distinct reaction mechanisms were calculated (Figure 1b and Supporting Information Figure S5). When trifluorotoluene was used as the substrate, the boryl anion initially attacked the electron‐deficient para position, resulting in a dearomative 1,4‐addition to form intermediate IM6. IM6 sequentially underwent defluoronation facilitated by * ^t^ *BuOBpin to give the neutral intermediate IM7. A tandem sequence of deprotonation by * ^t^ *BuOK, aromatization, and protonation occurred to yield the final product 4j. The nucleophilic attack of boryl anion on trifluorotoluene exhibited the highest energy barrier (24.0 kcal/mol), making it the rate‐determining step. A deuterium labeling experiment further supported the proposed mechanism. The reaction of 1a with 4‐D‐C_6_H_4_CF_3_ (**3j‐d_1_ **) afforded **4j‐d_1_ **, with the deuterium transferred to the difluoromethyl group in 90% deuteration rate, providing evidence of the reaction mechanism (Figure 1b).
For the α‐borylation of 4‐(trifluoromethyl)pyridine, although the exact mechanism remains unclear, a proposed reaction pathway has been provided, supported by a deuterium‐labeling experiment (Figure S5). The boryl anion IM2 preferentially attacked the most electron‐deficient ortho‐position, resulting in the formation of IM14. The deuterium labeling experiment with substrate **3l‐d_2_ ** [83] resulted in a 48% deuteration rate at the C5‐position of **4l‐d_2_ **, indicating the occurrence of a possible [1,4]‐hydrogen shift of IM14. Our calculations indicate that the coordination of 4‐(trifluoromethyl)pyridine to the −Bpin unit in IM2 (IM11) might facilitate the nucleophilic addition of boryl anion, reducing the energy barrier to 5.4 kcal/mol (TS6) (see Figure S5 for details).
Conclusion
3
In conclusion, a reactive NHC boryl anion was generated under mild conditions and applied as a highly nucleophilic boron synthon, exhibiting broad reactivity with various organic substrates. The neutral sp^2^–sp^3^ diboron reagent (NHC)BH_2_Bpin proved to be a versatile boron reagent and precursor in nucleophilic borylation reactions, enabling the discovery of unique reactivities that are difficult to achieve with traditional diboron reagents.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting File 1: The authors have cited additional references within the Supporting Information [84–98].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1E. J. Corey , “General Methods for the Construction of Complex Molecules,” Pure and Applied Chemistry 14 (1967): 19–38, 10.1351/pac 196714010019. · doi ↗
- 2E. J. Corey , “Robert Robinson Lecture. Retrosynthetic Thinking—Essentials and Examples,” Chemical Society Reviews 17 (1988): 111–133, 10.1039/CS 9881700111. · doi ↗
- 3C. Chen , J. Li , C. G. Daniliuc , C. Mück‐Lichtenfeld , G. Kehr , and G. Erker , “The [(NHC)B(H)C 6F 5] + Cations and Their [B](H)−CO Borane Carbonyls,” Angewandte Chemie International Edition 59 (2020): 21460–21464, 10.1002/anie.202009353.32705756 · doi ↗ · pubmed ↗
- 4T. S. De Vries , A. Prokofjevs , and E. Vedejs , “Cationic Tricoordinate Boron Intermediates: Borenium Chemistry From the Organic Perspective,” Chemical Reviews 112 (2012): 4246–4282, 10.1021/cr 200133 c.22519545 PMC 3394883 · doi ↗ · pubmed ↗
- 5J. M. Farrell , J. A. Hatnean , and D. W. Stephan , “Activation of Hydrogen and Hydrogenation Catalysis by a Borenium Cation,” Journal of the American Chemical Society 134 (2012): 15728–15731, 10.1021/ja 307995 f.22931196 · doi ↗ · pubmed ↗
- 6G. Kundu , P. R. Amrutha , S. Tothadi , and S. S. Sen , “Saturated NHC‐Stabilized Borenium, Boronium, Hydride‐Bridged Boron Cations, and a Bora‐Acyl Chloride,” Organometallics 43 (2024): 1355–1361, 10.1021/acs.organomet.4c 00139.
- 7Y. Ma , S.‐J. Lou , and Z. Hou , “Electron‐Deficient Boron‐Based Catalysts for C–H Bond Functionalisation,” Chemical Society Reviews 50 (2021): 1945–1967, 10.1039/D 0CS 00380 H.33325932 · doi ↗ · pubmed ↗
- 8T. Matsumoto and F. P. Gabbaï , “A Borenium Cation Stabilized by an N‐Heterocyclic Carbene Ligand,” Organometallics 28 (2009): 4252–4253, 10.1021/om 900476 g. · doi ↗