Proton Tautomerism for Anhydrous Superprotonic Conduction in 1,2,3‐Triazolium Dihydrogen Phosphate Crystal
Kaito Nishioka, Shun Dekura, Tomoko Fujino, Motohiro Mizuno, Reiji Kumai, Bo Thomsen, Motoyuki Shiga, Yuta Hori, Yasuteru Shigeta, Hatsumi Mori

TL;DR
This study shows how proton tautomerism can enable efficient proton conduction in a solid material without relying on molecular motion.
Contribution
The paper introduces proton tautomerism as a novel design principle for anhydrous proton conductors.
Findings
The 1,2,3-triazolium dihydrogen phosphate crystal shows isotropic superprotonic conductivity above 10−3 S cm−1.
Proton tautomerism enables low-barrier proton transport without molecular motion.
Both theoretical and experimental results confirm the role of tautomerism in proton conduction.
Abstract
Proton dynamics within molecular organic solids are crucial for energy‐related technologies. Proton conductors for use as solid electrolytes in hydrogen fuel cells have been developed, elucidating the higher proton transport mechanism and establishing design guidelines for higher conduction. Many anhydrous proton conductors for proton transport utilizing molecular motion in solids have been studied; however, low‐barrier conduction is challenging. In this study, we addressed proton tautomerism as a new guideline for proton conduction, rather than molecular motion. The key to facilitating low‐barrier conduction is proton transport without molecular motion via dynamic interconversion between multiple tautomers. We demonstrated the effectiveness of proton‐tautomerism strategy in 1,2,3‐triazole dihydrogen phosphate crystal, which exhibited low‐barrier, isotropic superprotonic conductivity…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
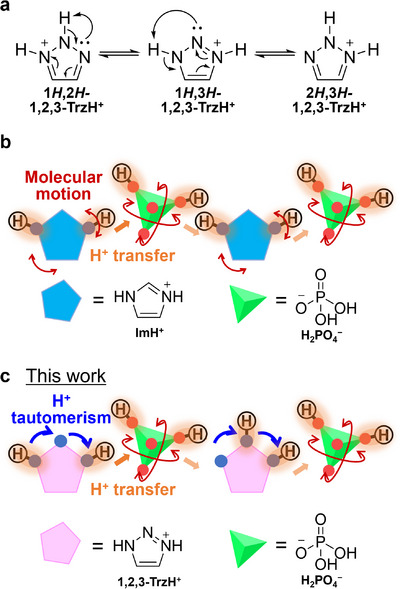 FIGURE 1
FIGURE 1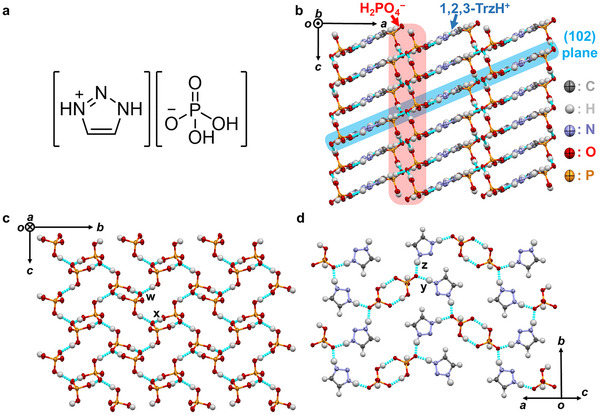 FIGURE 2
FIGURE 2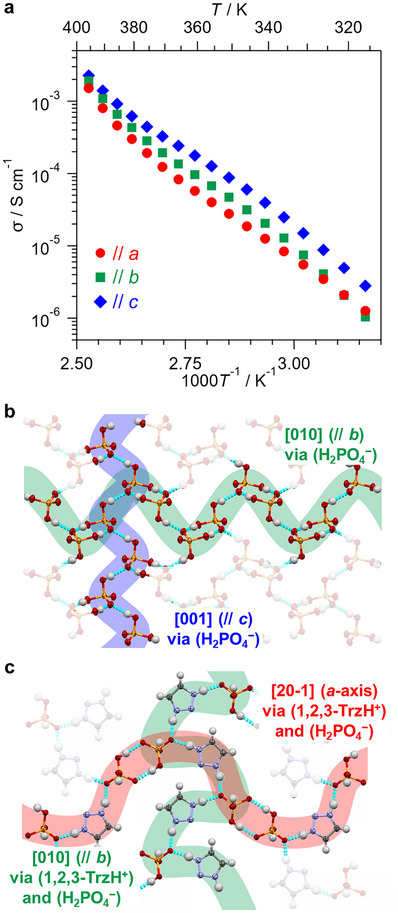 FIGURE 3
FIGURE 3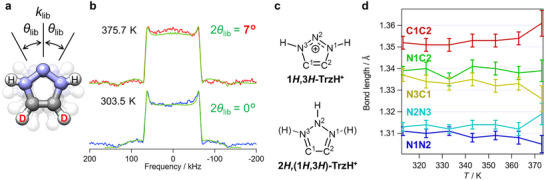 FIGURE 4
FIGURE 4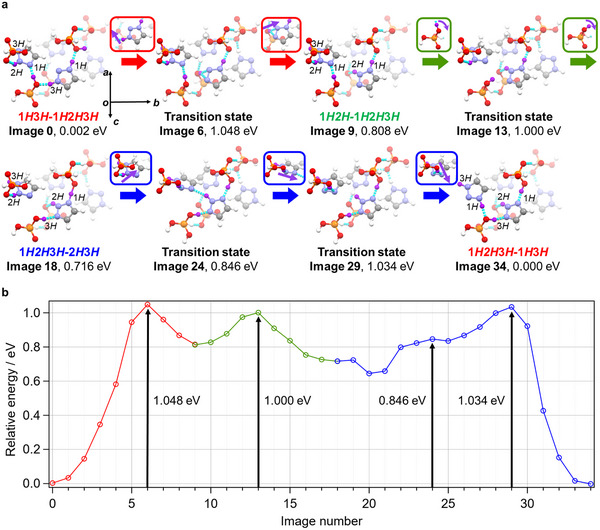 FIGURE 5
FIGURE 5| // | // | // | |
|---|---|---|---|
| σmax/S cm−1 |
1.51 × 10−3 [395.6 K] |
1.91 × 10−3 [395.6 K] |
2.26 × 10−3 [395.6 K] |
|
|
0.94(2) [316.0−395.6 K] |
0.97(1) [316.0−395.6 K] |
0.89(1) [316.0−395.6 K] |
- —Fellowship for Integrated Materials Science and Career Development and by JST SPRING
- —JSPS Grants‐in‐Aid for Scientific Research
- —JSPS Core‐to‐Core Program
- —MEXT Grants‐in‐Aid for Scientific Research on Innovative Areas “Hydrogenomics”
- —Nanotechnology Platform Program <Molecule and Material Synthesis>
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFuel Cells and Related Materials · Organic and Molecular Conductors Research · Advanced battery technologies research
Introduction
1
Control of proton dynamics in molecular organic solids is of fundamental scientific interest and essential for anhydrous high‐proton conductors, that is, solid electrolytes for hydrogen fuel cells operable in the medium temperature range above 100°C with no need for humidified environments [1, 2, 3]. However, issues such as dopant leakage, swelling‐induced degradation, and reaction with electrodes remain. To address these problems while maintaining high conductivity and realizing excellent conductors, new solid electrolytes must be developed. A material design guideline based on an understanding of the conduction mechanism is essential. However, given the nonstoichiometric and heterogeneous compositions of doped polymers and porous materials, and their lack of crystallinity, exploration of conduction mechanisms based on structure–property relationships is extremely difficult.
Intrinsic anhydrous proton conductivity has recently been studied using single‐crystal samples to establish design guidelines for achieving high conductivity [3, 4, 5, 6, 7, 8, 9, 10, 11]. Molecular organic single crystals are considered ideal model materials because they have well‐defined structures, and the conduction mechanism is not influenced by interfaces and defects. Previous studies on acid–base‐type imidazolium dicarboxylate single crystals suggested that the anhydrous proton‐conduction mechanism in molecular crystals is based on a Grotthus mechanism [12]; in other words, the conduction mechanism is based on a hydrogen bond network structure involved in a conduction pathway, intermolecular differences in acidity for intermolecular proton transfer, and molecular reorientation preceding intramolecular proton transfer from one side to the other side within one molecule [7, 8]. Therefore, proton conductors for superprotonic conductivity (>10^−4^ S cm^−1^) have been developed based on design strategies of utilizing molecular motion. Although realization of superprotonic conductivity has been reported [3, 6, 11], systematic control of molecular motion in crystals has not been established. Molecular motion‐induced proton conduction has been achieved; nonetheless, in most cases, the high activation energies (E a about 2−6 eV) would prevent conductivity enhancement [7, 8, 9, 10]. Therefore, an intramolecular proton transfer mechanism that can be strategically introduced and controllable at the molecular design stage is necessary as an alternative to molecular reorientation.
Proton tautomerism is a phenomenon in which isomers with different intramolecular proton positions undergo rapid interconversion in conjunction with recombination of the π‐electronic structures (Figure 1a). Generally, proton tautomerism is known in organic chemistry as a peculiar molecular property [13, 14, 15]; however, few studies have focused on proton tautomerism in crystals. In conventional conduction mechanisms such as that of imidazolium dihydrogen phosphate (**ImH^+^ **)(H_2_PO_4_ ^−^) [16], when a molecule receives a proton from its left neighbor, molecular rotation (for intramolecular proton transfer) is necessary to pass it to its right neighbor (Figure 1b). However, reorientation‐free intramolecular proton transfer is possible for a molecule exhibiting proton tautomerism, such as 1,2,3‐triazolium (=**1,2,3‐TrzH^+^ **; Figure 1a). Unlike molecular motion, proton tautomerism can be incorporated at the molecular design stage, its conduction mechanism can be discussed based on the static average structure, and strategic achievement of low‐barrier long‐distance proton transfer by controlling the molecular arrangement is likely possible. Previous studies reported anhydrous proton conduction in materials containing multiple tautomeric species, particularly 1,2,3‐Trz, where E a was low (∼0.3 eV) [17, 18, 19, 20, 21, 22, 23]. However, in all of the previous studies, the specific role of multiple tautomers in the solid‐state proton conduction remains unexplored experimentally, and the dynamic interconversion of tautomers—which is, in essence, proton tautomerism—has not been demonstrated experimentally to contribute directly to proton conduction. Moreover, the solid‐state proton conductivity of 1,2,3‐Trz was only ∼3 × 10^−5^ S cm^−1^ because of the poor thermal stability of the neutral crystal. Therefore, proton tautomerism has never been conclusively proven to directly govern proton conduction and has not been considered a practical guideline for realizing high proton conductivity.
*Proton tautomerism and proton conduction based on molecular motion and proton tautomerism. (a) Changes in chemical structures and π‐electronic configuration of 1,2,3‐TrzH+ . (b) Rotational motion of ImH
and H2PO4 −. (c) Proton tautomerism of 1,2,3‐TrzH
- and rotational motion of dihydrogen H2PO4 −.*
This study realized isotropic superprotonic conduction exceeding 10^−3^ S cm^−1^ using proton tautomerism in acid–base single crystals and elucidated the contribution of proton tautomerism to solid‐state proton conduction for the first time. For comparison with a previous study on imidazolium phosphate [16], we consider phosphoric acid and 1,2,3‐Trz as acid and base molecules, respectively; the former and latter conduct protons through well‐understood isotropic reorientation [24, 25, 26, 27, 28, 29] and unexplored proton tautomerism in a solid, respectively (Figure 1c). Hence, we prepare new single crystals of 1,2,3‐triazolium dihydrogen phosphate (**1,2,3‐TrzH^+^ **)(H_2_PO_4_ ^−^) (=1). Despite the anisotropic hydrogen‐bonding network of the crystal structure, an isotropic anhydrous superprotonic conductivity exceeding 10^−3^ S cm^−1^ is achieved at 396 K. This is the first instance of an anhydrous crystal exceeding 10^−3^ S cm^−1^ facilitated by proton tautomerism. Solid‐state ^2^H nuclear magnetic resonance (NMR) spectroscopy using **1,2,3‐TrzH^+^ ** partially H/D isotopically substituted (**1,2,3‐Trz‐d 2‐H^+^ **)(H_2_PO_4_ ^−^) polycrystals suggests that **1,2,3‐Trz‐d 2‐H^+^ ** exhibits minor libration motion that does not contribute to conduction. Variable‐temperature single‐crystal x‐ray structural analysis, ab initio nonequilibrium molecular dynamics (NEMD), and density functional theory (DFT) calculations confirm for the first time that the **1,2,3‐TrzH^+^ ** proton tautomerism can aid the development of superprotonic conductivity in solids.
Results and Discussion
2
Thermal Properties
2.1
High‐quality single crystals of 1 were newly obtained through dichloromethane vapor diffusion from a mixture of 1,2,3‐Trz and phosphoric acid in a 1:1 stoichiometric ratio in acetone (see the Supporting Information for details). The thermal stability of these single crystals was evaluated based on melting‐point measurement and thermogravimetry/differential thermal analysis (TG/DTA). Melting occurred at 410 K: an endothermic peak and weight loss were concomitantly observed from the DTA and TG chart, respectively. Thus, sample decomposition, possibly 1,2,3‐Trz evaporation, occurred at approximately the melting point (Figure S7). The phase behavior of single‐crystalline 1 was investigated using differential scanning calorimetry (DSC) below the melting point. From room temperature to 400 K, the DSC chart exhibited no peaks indicative of phase transitions. Overall, 1 was stable in the mid‐temperature range above 100°C (373 K) to ∼400 K with no phase transition. These characteristics support the application of 1 as an anhydrous solid electrolyte for fuel cells.
Single‐Crystal Structure of 1
2.2
X‐ray structural analysis at 298 K (Figure 2) revealed the crystal structure of 1 [30]. The space group was monoclinic P2_1_/c with crystallographically independent one **1,2,3‐TrzH^+^ ** and one H_2_PO_4_ ^−^ molecules in a 1:1 stoichiometric ratio. The unit cell contained four pairs of (**1,2,3‐TrzH^+^ **)(H_2_PO_4_ ^−^). The crystals were anhydrous: free of water and other crystallization solvent molecules (Figure 2a and Table S1). H_2_PO_4_ ^−^ anions formed a two‐dimensional (2D) hydrogen‐bonding network extending on the bc plane with hydrogen‐bond distances d O···O = 2.573(2), 2.586(2) Å (Figure 2c). From the hydrogen‐bond distances, the hydrogen‐bond protons between the H_2_PO_4_ ^−^ anions were in double‐well potentials. No disorder of the hydrogen‐bonded protons was observed. The **1,2,3‐TrzH^+^ ** cations were in the 2D H_2_PO_4_ ^−^‐layer stacking direction (//a), and the H_2_PO_4_ ^−^ and **1,2,3‐TrzH^+^ ** layers alternated in the a‐axis direction (Figure 2b). In contrast to the H_2_PO_4_ ^−^ layer, no hydrogen bonds existed between the **1,2,3‐TrzH^+^ ** cations in the **1,2,3‐TrzH^+^ ** layers. Hydrogen bonds were present between the H_2_PO_4_ ^−^ and **1,2,3‐TrzH^+^ ** layers, with distances d N···O = 2.609(3), 2.661(3) Å, forming a hydrogen‐bond network extending in the (102) plane (Figure 2d). Overall, 1 featured a 3D hydrogen‐bonding network comprising anisotropic molecular arrangements; this is likely an anhydrous proton‐conductive structure in any crystal axial direction.
Chemical and crystal structures of 1,2,3‐triazolium dihydrogen phosphate (=1). (a) Chemical structure of 1. (b, c) Novel crystal structure of 1 along b‐ and a‐axes, respectively (w, x: hydrogen bonds of d O···O = 2.573(2), 2.586(2) Å). (d) (102) plane in crystal structure (y, z: hydrogen bonds of d N···O = 2.609(3), 2.661(3) Å). Gray: C, white: H, blue: N, red: O, orange: P. The light‐blue dashed lines correspond to hydrogen bonds.
Single‐Crystal Anhydrous Proton Conductivity of 1
2.3
To elucidate the intrinsic anhydrous proton conductivity of 1 and its anisotropy, the temperature dependence of anhydrous proton conductivity was measured along each crystallographic axis using alternating‐current (AC) impedance spectroscopy and single‐crystalline samples (see the Supporting Information for details). The crystals were covered with a heat‐resistant epoxy resin to prevent 1,2,3‐Trz evaporation from the surface during measurement (Figure S9), and the proton conductivity (σ) along each axis was evaluated as a function of temperature to ∼400 K (Figure 3a). In all directions, 1 exhibited superprotonic conductivity exceeding 10^−3^ S cm^−1^ at 395.6 K. Protons (H^+^) were identified as the conducting carriers using a fuel‐cell device with single crystals of 1 (Figures S3 and S19). Despite the anisotropic molecular arrangement (Figure 2), the conductivity anisotropy was low. The E a estimated from the temperature dependence of σ were 0.94(2), 0.98(1), and 0.89(1) eV along the a, b, and c axes, respectively: all <1 eV (Table 1). These E a values are considerably lower than those of single‐crystalline salts based on the rotational motions of **ImH^+^ ** cations (∼2–6 eV)^16^, and comparable to those of other reported phosphate proton conductors [5, 6].
Temperature dependence of anhydrous proton conductivity (σ) of 1 and possible conduction pathways. (a) Red circles: along a‐axis, green squares: along b‐axis, blue diamonds: along c‐axis. (b) Pathways along b‐ and c‐axes in 2D H2PO4 − layer in (100) plane. (c) Pathways along b‐axis and [20−1] in (102) plane (gray: C, white: H, blue: N, red: O, orange: P).
TABLE 1: Maximum anhydrous proton conductivity, σmax, and activation energy, E a, of single‐crystal samples of 1 for each crystallographic direction.
As shown in Figure 2, a 2D hydrogen bonding network of H_2_PO_4_ ^−^ anions extends in the bc plane. In the b‐ and c‐axis directions, possible proton‐conduction pathways traversed H_2_PO_4_ ^−^ only (Figure 3b). Because **1,2,3‐TrzH^+^ ** exists between H_2_PO_4_ ^−^ in the (102) plane, an alternative proton‐conduction pathway along the b‐axis is possible, via both **1,2,3‐TrzH^+^ ** and H_2_PO_4_ ^−^ (Figure 3c; green). However, proton conduction along the a‐axis is possible only via both **1,2,3‐TrzH^+^ ** and H_2_PO_4_ ^−^ (Figure 3c; red). Although the conduction pathways along the respective crystal axes differ (i.e., they are anisotropic), the experimentally observed conductivities and their E a values are isotropic (Figure 3a and Table 1). With the isotropic and low E a values, these results indicate that proton conduction through **1,2,3‐TrzH^+^ ** cations is as fast and low‐barrier as that through H_2_PO_4_ ^−^ anions, possibly due to the proton tautomerism of the **1,2,3‐TrzH^+^ ** cations. To the best of our knowledge, this is the first example of superprotonic conductivity exceeding 10^−3^ S cm^−1^ in anhydrous crystals exhibiting proton tautomerism.
Molecular Motions of 1,2,3‐TrzH+
From Variable‐Temperature Solid‐State 2H NMR
2.4
Although the H_2_PO_4_ ^−^ rotational dynamics contribute to proton conduction, fast and low‐barrier proton conduction via **1,2,3‐TrzH^+^ ** is possible based on proton tautomerism and molecular rotational motions. To investigate the molecular rotational dynamics of **1,2,3‐TrzH^+^ ** cations in 1, we performed variable‐temperature solid‐state ^2^H NMR measurements using a polycrystalline sample of 1. Solid‐state ^2^H NMR enables investigation of kilohertz‐ to megahertz‐order dynamics, facilitating the evaluation of molecular rotational dynamics in crystals [11, 31, 32]. Here, two C–H protons of **1,2,3‐TrzH^+^ ** were H/D substituted (=**1,2,3‐Trz‐d 2‐H^+^ **; Figure 4a) for selective observation of the cation dynamics only; this did not influence the thermal, structural, and physical properties (Tables S1 and S2, Figures S7 and S15−S18) [30]. Figure 4b shows the solid‐state ^2^H NMR spectra at 303.5 and 375.5 K; with heating, the spectral shape changed only minimally. Spectral simulation assuming librational motion with librational frequency k lib and angle θ lib reproduced the experimental spectra (Figures 4a andS4). No librational motion was indicated at 303.5 K. Minor librational motion occurred at 375.7 K with k lib of 10^8^ Hz and 2θ lib of 7°. Although the anhydrous proton conductivity increased by approximately three orders of magnitude, from below 10^−6^ to ∼10^−3^ S cm^−1^, 2θ lib was only 7° at 375.5 K, suggesting little molecular motion. In our previous work, **Im‐d 3‐H^+^ ** cations in (**Im‐d 3‐H^+^ **)(H_2_PO_4_ ^−^) salt exhibited librational motion with 2θ lib = 46° at a maximum temperature of 357 K; the conductivity reached ∼10^−5^ S cm^−1^. [16] In contrast, the **1,2,3‐Trz‐d 2‐H^+^ ** cations in **1‐d 2 ** had considerably lower kinetics with conductivity more than two orders higher. As evident from the NMR results, the rotational (librational) dynamics of **1,2,3‐TrzH^+^ ** contribute minimally to the high conductivity in 1, with the fast proton conduction through **1,2,3‐TrzH^+^ ** being achieved via proton tautomerism.
Solid‐state 2H NMR of 1‐d 2 and variable‐temperature XRD of 1. (a) Model of librational motion of 1,2,3‐TrzH+ assumed in simulation of solid‐state 2H NMR spectra (gray: C, white: H, or D, blue: N). (b) Temperature dependence of 2H NMR spectra for 1‐d 2 . The green lines show the simulated powder patterns assuming libration of 1,2,3‐TrzH+ . Fixed values of e 2 qQ/ħ = 175 kHz and η = 0.04 were used for the simulation (see Supporting Information for details). (c) Chemical structures and corresponding atomic labels of 1H,3H‐TrzH+ and 2H,(1H,3H)‐TrzH+ . (d) Temperature dependence of bond lengths of 1,2,3‐TrzH+ in 1. The error bars correspond to the estimated standard deviations.
Temperature Dependence of the Bond Lengths in 1,2,3‐TrzH+
2.5
To obtain direct experimental evidence of the proton tautomerism of **1,2,3‐TrzH^+^ ** cations in 1, we performed variable‐temperature single‐crystal X‐ray structural analyses at 313–373 K. [30] The crystal system and space groups were temperature‐independent, consistent with the peak‐free DSC result; the crystal axis length and unit cell volume increased monotonically (Table S3). The room‐temperature structural analysis suggested that **1H,3H‐TrzH^+^ ** was the most stable tautomer in 1 (Figure 2); thus, thermally activated protons will appear at the 2H site of the **1,2,3‐TrzH^+^ ** cations at high temperature under proton tautomerism (Figure 4c). However, no 2H protons were observed in the high‐temperature structures, possibly due to the low sensitivity of x‐ray to hydrogen and the low ratio of the 2H‐tautomer. The tautomer ratio was alternatively evaluated from the bond lengths of the **1,2,3‐TrzH^+^ ** five‐membered ring, because the proton tautomerism involves π‐electron reconstruction (Figure 4c).
The theoretical bond‐length changes for the **1,2,3‐TrzH^+^ ** proton tautomerism were estimated using DFT calculations. The N^1^–N^2^, N^2^–N^3^, and C^1^–C^2^ bonds elongate, and the N^1^–C^2^ and N^3^–C^1^ bonds contract when the **1H,3H‐TrzH^+^ ** tautomer converts to the **2H,(1H,3H)‐TrzH^+^ ** tautomer via proton tautomerism (Figure S20; see also the Experimental section in the Supporting Information). Hence, we experimentally investigated the temperature dependence of the bond lengths in the **1,2,3‐TrzH^+^ ** five‐membered ring. Evident temperature dependencies of the bond‐length changes were noted for N^2^–N^3^, N^3^–C^1^, and C^1^–C^2^, with N^2^–N^3^ and C^1^–C^2^ extending and N^3^–C^1^ detracting with increasing temperature (Figure 4d). However, N^1^−N^2^ exhibited counterintuitive behavior. These changes correspond to the theoretically predicted increase in the **2H,(1H,3H)‐TrzH^+^ ** proportion due to proton tautomerism. Therefore, an increased fraction of **2H,(1H,3H)‐TrzH^+^ ** tautomer in the single crystal of 1 with temperature was observed experimentally, suggesting that proton tautomerism contributes to the proton‐conduction mechanism in 1.
Ab Initio Calculations for Proton Conduction in 1
2.6
To investigate the responsibility of **1,2,3‐TrzH^+^ ** proton tautomerism for proton conduction, we performed ab initio NEMD simulations (Figures S22–S24). No molecular rotational motion or tautomerism of **1,2,3‐TrzH^+^ ** was observed for simulations of a perfect crystal; however, proton transfer with H_2_PO_4_ ^−^ rotation was observed for models with excess protons or proton defects. No large‐angle rotational motion of **1,2,3‐TrzH^+^ ** was observed, even for those doped models. The calculated perfect‐crystal proton conductivity was ∼10^−5^ S cm^−1^ in all three axial directions (see the Experimental section in the Supporting Information for details); the conductivity of the crystal with excess protons or proton defects was ∼10^−3^ S cm^−1^ in all three axial directions, agreeing with experimental AC impedance measurements. Thus, proton conduction likely occurs near proton excess or defect sites rather than in perfect crystals.
To elucidate the proton‐conduction pathway involving proton tautomerism in **1,2,3‐TrzH^+^ ** and its E a, nudged elastic band (NEB) method using DFT calculations was performed. The proton conduction was in the −b‐axis direction, along which proton conduction through **1,2,3‐TrzH^+^ ** proton tautomerism and intermolecular proton transfer without H_2_PO_4_ ^−^ rotational motion is possible. In the NEB calculations, we constructed three structural models: a‐ perfect crystal and crystals with one proton excess and one defect site introduced in the unit cell, respectively. Two proton‐conduction pathways were found in perfect crystals (Figures S25 and S26), with E a ≈ 2 eV, significantly exceeding the experimentally estimated E a (∼1 eV). This suggests that the presence of excess or deficient protons is necessary, consistent with the NEMD simulations.
The relative energy diagram obtained from NEB calculations for the models with proton excess or deficient sites reproduced the experimental E a values, unlike the perfect‐crystal case. Figure 5a shows the suggested conduction pathway along the −b direction based on the obtained minimum‐energy paths for the proton excess‐site case. We first assumed the conduction pathway along the −b direction through four types of metastable structures and optimized these structures independently under the periodic boundary condition of the single unit cell. The structure containing 2H‐1,2,3‐Trz with a proton on the 2H position (**1H3H–1H2H3H **; the label does not indicate which proton is attached between the acid or base molecules) was the initial structure, followed by **1H2H–1H2H3H ** and **1H2H3H–2H3H **, to the final state of **1H2H3H–1H3H **. The proton was transferred to the crystallographically equivalent **1,2,3‐TrzH^+^ **; this can be repeated to produce a long‐range conduction pathway. The optimized metastable structures were used as the initial or final states. The intermediate states and their relative energies were determined by three NEB calculation processes as the minimum energy paths. The intermediate structures obtained from the NEB calculations (Images 0–34, Figure 5a) visualized the tautomerization process from the initial to final state. Interestingly, the **1,2,3‐TrzH^+^ ** tautomerization was achieved via transient proton transfer from **1,2,3‐TrzH^+^ ** to H_2_PO_4_ ^−^ in the adjacent layer (Images 6, 24, and 29, Figure 5a). The protons then returned from H_3_PO_4_ to 1,2,3‐Trz, and proton transfer occurred from **1H,2H‐TrzH^+^ ** to H_2_PO_4_ ^−^, forming 2H‐1,2,3‐Trz (Images 9 and 34, Figure 5a). The estimated E a values of the proton conduction along the −b direction involving the **1,2,3‐TrzH^+^ ** proton tautomerism were estimated to be 1.048 eV (Figure 5b), consistent with experimental results (∼1 eV; Table 1). Similarly, for the structural model with proton defects, the E a of the **1,2,3‐TrzH^+^ ** proton tautomerism was consistent with the experimental value (Figure S27). The **1,2,3‐TrzH^+^ ** proton tautomerism requires H_2_PO_4_ ^−^ assistance because of the high barrier for a single molecule in vacuum (∼2.5 eV; Figure S21). Thus, the super‐protonic conduction in 1 realized in this study is specific to a condensed matter system; the crystal environment (e.g., the molecular arrangements and hydrogen‐bonding network structures) is important. These calculations strongly indicate that the fast proton conduction in 1 is realized through **1,2,3‐TrzH^+^ ** proton tautomerism.
Ab initio NEB calculation of proton tautomerism of 1. (a) Structures of local minimum states and transition states (gray: C, white: H, blue: N, red: O, orange: P; the conducting protons are colored purple). NEB calculations were performed for each of the three proton transfer divisions. Images 0, 9, 18, and 34 were optimized individually and correspond to the initial or final states of the respective proton transfer divisions. Images 6, 13, 24, and 29 are the transition states. The corresponding image numbers and relative potential energies (the lowest value for Image 34 was set to 0) are shown beneath the structures. (b) Relative energy curve obtained from NEB calculation for tautomerism process of 1 (red, green, and blue: the first, second, and third divisions, respectively).
Conclusion
3
In 1, two possible proton‐conduction pathways exist: one through H_2_PO_4_ ^−^ alone and the other through both **1,2,3‐TrzH^+^ ** and H_2_PO_4_ ^−^. The proton conduction through H_2_PO_4_ ^−^ is well understood, occurring via H_2_PO_4_ ^−^‐anion reorientational motion [25, 26, 27]. The latter alternative can occur via the pathways proposed by our theoretical calculations (Figures 5 and S27). Such proton conduction comprises the following steps: 1) hydrogen‐bond reconstruction with H_2_PO_4_ ^−^ rotation; 2) intermolecular proton transfer between H_2_PO_4_ ^−^ and **1,2,3‐TrzH^+^ **; and 3) **1,2,3‐TrzH^+^ ** proton tautomerism assisted by H_2_PO_4_ ^−^ in the adjacent (102) layer (Figure 5a). Proton conduction via proton tautomerism is supported by the following findings: The overall **1,2,3‐TrzH^+^ ** molecular motion revealed by solid‐state ^2^H NMR was almost zero, the temperature dependence of the bond lengths revealed changes in proportion of the tautomers with temperature, and the NEB‐calculated E a assuming proton tautomerism of **1,2,3‐TrzH^+^ ** was up to 1.048 eV. These findings distinguish this work from previous reports [17, 18, 19, 20, 21, 22, 23], in which the dynamic interconversion of tautomers had not been experimentally verified.
It is worth nothing that, in this study, the proton conductivity is analyzed assuming Arrhenius behavior. Some crystalline materials involving molecular motions are known to exhibit non‐Arrhenius behavior [8, 11, 16]. Nevertheless, in 1, the temperature dependence of the proton conductivity looks follow Arrhenius behavior, and the E a determined from the Arrhenius fit is consistent with the E a estimated by first‐principles calculations. Thus, an Arrhenius‐type analysis is justified in this study. Still, we cannot conclude that such a new conduction mechanism in this system is physically interpreted with an Arrhenius equation, which remains an issue for future investigation.
The combined experimental and theoretical results indicate that the isotropic anhydrous superprotonic conduction in 1 primarily stems from proton tautomerism, in addition to the reorientational motion of phosphate anions. Because proton conduction can occur without involving the entire molecular motion, low‐barrier conduction becomes possible. Moreover, changes in the electronic structure associated with proton tautomerism are observable as bond‐length variations. Thus, proton tautomerism can facilitate a new development strategy for efficient anhydrous superprotonic conductors.
For the first time, we experimentally confirmed that the microscopic proton tautomerism in a single molecule can induce a macroscopic crystalline property, namely, superprotonic conduction. Combining the conventional design guideline of increasing molecular motion with thermal‐stability improvement is challenging; however, proton tautomerism can be exploited with effective material design for thermal‐stability improvement; for example, by increasing molecular weights. Therefore, proton tautomerism is advantageous for the realization of highly proton‐conductive materials operable at high temperatures. Use of proton tautomerism in not only molecular crystals but also metal complex crystals and crystalline polymers is expected to enable the production of outstanding energy materials.
Author Contributions
K.N., S.D., and H.M. conceived the idea and designed the study. K.N. performed the experiments and the NEB calculations and analyzed the results. T.F. helped the synthesis of **1,2,3‐Trz‐d 2 **. M.M. developed the simulation software for the solid‐state ^2^H NMR spectra. R.K. contributed to the XRD experiments by advising on their design and interpretation. B.T. and M.S. performed the NEMD calculations and analyzed the results. Y.H. and Y.S. contributed to the NEB calculations by advising on their design and interpretation. K.N., S.D., and H.M. wrote and refined the paper. H.M. supervised the project. All authors discussed the results.
Conflicts of Interest
The authors declare no conflict of interest.
Supporting information
Supporting File 1: The authors have cited additional references within the Supporting Information [1–27].
Supporting File 2: anie71356‐sup‐0002‐Data.zip.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1S. Bureekaew , S. Horike , M. Higuchi , et al., “One‐Dimensional Imidazole Aggregate in Aluminium Porous Coordination Polymers With High Proton Conductivity,” Nature Materials 8 (2009): 831–836, 10.1038/nmat 2526.19734885 · doi ↗ · pubmed ↗
- 2H. Gao and K. Lian , “Proton‐Conducting Polymer Electrolytes and Their Applications in Solid Supercapacitors: A Review,” RSC Advances 4 (2014): 33091–33113, 10.1039/C 4RA 05151 C. · doi ↗
- 3R. M. P. Colodrero , P. Olivera‐Pastor , A. Cabeza , and M. Bazaga‐García , “Properties and Applications of Metal Phosphates and Pyrophosphates as Proton Conductors,” Materials 15 (2022): 1292, 10.3390/ma 15041292.35207833 PMC 8875660 · doi ↗ · pubmed ↗
- 4A. I. Baranov , V. P. Khiznichenko , and L. A. Shuvalov , “High Temperature Phase Transitions and Proton Conductivity in Some KDP‐Family Crystals,” Ferroelectrics 100 (1989): 135–141, 10.1080/00150198908007907. · doi ↗
- 5Y. Yoshii , N. Hoshino , T. Takeda , and T. Akutagawa , “Protonic Conductivity and Hydrogen Bonds in (Haloanilinium)(H 2PO 4) Crystals,” Journal of Physical Chemistry C 119 (2015): 20845–20854.
- 6G. Yuan , T. Takeda , N. Hoshino , and T. Akutagawa , “Highly Proton‐Conducting Mixed Proton‐Transferred [(H 2PO 4–)(H 3PO 4)]∞ Networks Supported by 2,2′‐Diaminobithiazolium in Crystals,” Journal of Physical Chemistry C 124 (2020): 1861–1871.
- 7Y. Sunairi , A. Ueda , J. Yoshida , K. Suzuki , and H. Mori , “Anisotropic Proton Conductivity Arising From Hydrogen‐Bond Patterns in Anhydrous Organic Single Crystals, Imidazolium Carboxylates,” Journal of Physical Chemistry C 122 (2018): 11623–11632, 10.1021/acs.jpcc.8b 00814. · doi ↗
- 8Y. Sunairi , S. Dekura , A. Ueda , T. Ida , M. Mizuno , and H. Mori , “Anhydrous Purely Organic Solid‐State Proton Conductors: Effects of Molecular Dynamics on the Proton Conductivity of Imidazolium Hydrogen Dicarboxylates,” Journal of the Physical Society of Japan 89 (2020): 051008, 10.7566/JPSJ.89.051008. · doi ↗