A Case of a Novel Perforin Gene Variant in Severe Familial Hemophagocytic Lymphohistiocytosis Type 2 (FHL2)
Hiroshi Yamauchi, Moeko Hino, Kazuyuki Meguro, Taiji Nakano, Takahiro Aoki, Yoshiharu Yamashita, Tomoko Okunushi, Takeshi Yamamoto, Hironori Sato, Takahiro Yasumi, Yuiko Hirata, Hirofumi Shibata, Hiroshi Nakajima, Hiromichi Hamada

TL;DR
A 5-month-old boy with severe FHL2 was found to have a new PRF1 gene variant, A21V, which contributes to the disease when combined with another variant.
Contribution
The study reports a novel PRF1 variant, A21V, and provides functional evidence of its pathogenicity in FHL2.
Findings
The PRF1 A21V variant is novel and associated with reduced perforin expression in NK cells.
The A21V/P16S compound heterozygous variant combination contributes to early-onset FHL2.
Functional validation supports the pathogenic role of the P16S variant in the context of A21V.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a life‐threatening hyperinflammatory syndrome caused by excessive cytokine release from activated T cells and macrophages. Primary HLH, or familial HLH (FHL), results from genetic mutations affecting cytotoxic lymphocyte function. We present a case of FHL Type 2 (FHL2) caused by compound heterozygous variants in the PRF1 gene, including one novel missense variant of p.Ala21Val (A21V). A 5‐month‐old boy presented with persistent fever, pancytopenia, coagulopathy, hepatosplenomegaly, and elevated ferritin, meeting the HLH‐2004 diagnostic criteria. Bone marrow revealed hemophagocytosis, and NK cell activity was markedly reduced. Genetic analysis identified compound heterozygous PRF1 variants: A21V and p.Pro16Ser (P16S). Flow cytometric analysis demonstrated markedly reduced PRF1 protein expression in the patient’s NK cells. The patient was…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
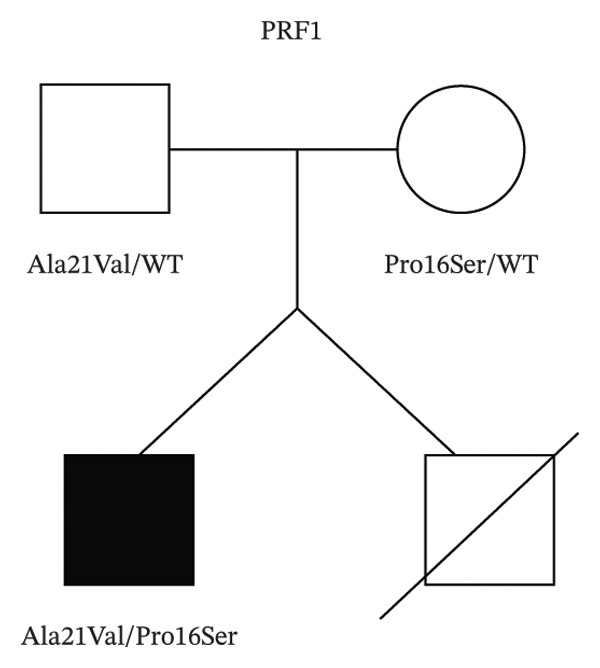 Figure 1
Figure 1 Figure 2
Figure 2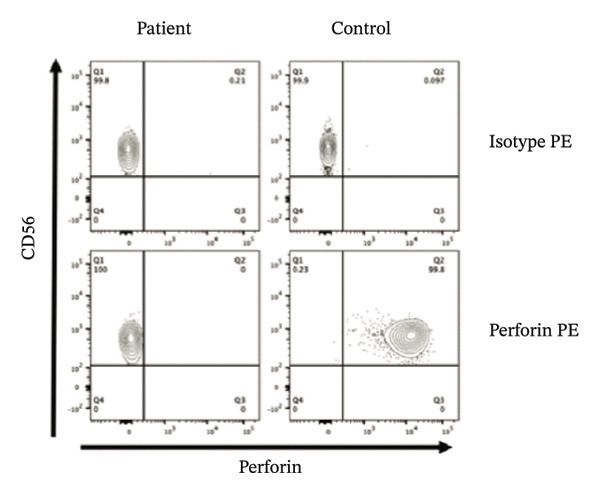 Figure 3
Figure 3| Parameter | Result | Normal range (if available) |
|---|---|---|
| WBC | 2700/μL | 5000–10,000/μL |
| RBC | 268 × 104/μL | 410–530 × 104/μL |
| Hb | 7.2 g/dL | 13.0–17.0 g/dL |
| Plt | 1.1 × 104/μL | 13–35 × 104/μL |
| Band | 1.5% | 0%–5% |
| Seg | 0% | 40%–70% |
| Eos | 0% | 1%–4% |
| Baso | 0% | 0%–1% |
| Mono | 3.5% | 2%–8% |
| Lymph | 78% | 20%–40% |
| Reac‐Lymph | 17% | 0%–5% |
| TP | 3.8 g/dL | 6.5–8.0 g/dL |
| Alb | 2.4 g/dL | 4.0–5.0 g/dL |
| CK | 665 U/L | 45–163 U/L |
| AST | 367 U/L | 13–30 U/L |
| ALT | 267 U/L | 10–42 U/L |
| LD | 1477 U/L | 120–240 U/L |
| Cre | 0.2 mg/dL | 0.6–1.1 mg/dL |
| UN | 6 ng/dL | 8–20 mg/dL |
| TG | 288 mg/dL | < 150 mg/dL |
| Na | 130 mmol/L | 135–145 mmol/L |
| K | 3.5 mmol/L | 3.6–5.0 mmol/L |
| CRP | 1.18 mg/dL | < 0.3 mg/dL |
| FER | 5403 ng/mL | 20–200 ng/mL |
| APTT | 32.4 s | 25–35 s |
| PT‐INR | 1.9 | 0.9–1.1 |
| Fib | 51 mg/dL | 200–400 mg/dL |
| FDP | 6 mg/mL | < 5 μg/mL |
| D‐dimer | 2.7 mg/mL | < 1.0 μg/mL |
| AT‐III | 44% | 80%–120% |
| IgG | 214 mg/dL | 861–1747 mg/dL |
| C3 | 92 mg/dL | 79–152 mg/dL |
| C4 | 28 mg/dL | 16–38 mg/dL |
| sIL‐2R | 25,995 U/mL | 121–613 U/mL |
| BM‐NCC | 48,000/μL | 10,000–20,000/μL |
| BM‐MgK | 12/μL | 2–10/μL |
| BM‐M/E | 2.1 | 1.5–3.3 |
| NK cell activity | 8% | 18%–40% |
| CMV‐IgM | 0.2 (−) | Negative |
| EBV‐IgM | 0.2 (−) | Negative |
| HSV‐IgM | 0.2 (−) | Negative |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Immune Cell Function and Interaction · Otitis Media and Relapsing Polychondritis
1. Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a fatal disease characterized by excessive inflammatory cytokine production [1]. Primary HLH is caused by genetic factors, and multiple causative variants have been identified [2–10]. The PRF1 gene encodes perforin, a major component of cytotoxic granules. Abnormal PRF1 is an important factor in primary HLH [2–5, 9, 10]. Although numerous PRF1 variants have been reported, interpretation of rare missense variants remains challenging, especially when the variant is unreported or functional evidence is lacking. Here, we report a patient with severe early‐onset familial HLH (FHL) Type 2 (FHL2) caused by compound heterozygous PRF1 variants, including one novel missense variant. This case highlights the importance of integrating genomic and functional analyses to clarify the pathogenicity of rare PRF1 variants.
2. Case Presentation
A 5‐month‐old boy was admitted with a chief complaint of fever. He was born at 38 weeks and 5 days of gestation without perinatal complications, with a birth weight of 3062 g. He was the first child of nonconsanguineous parents and had a twin sibling who had died in utero at 6 months of gestation.
Five days before admission, the patient developed a fever accompanied by malfeeding. Two days before admission, his condition worsened, with frequent watery stools and progressive lethargy, suggesting a systemic inflammatory response. His previous physician suspected HLH caused by Epstein–Barr virus (EBV) infection and referred him to our hospital.
On admission, body temperature was 38.6°C, blood pressure 95/50 mmHg, and oxygen saturation 95% on nasal cannula oxygen at 0.5 L/min. He appeared lethargic with poor vitality. His skin showed reticular, erythematous patches throughout the body. Breath sounds were markedly diminished bilaterally. The liver was palpable 3.0 cm below the right costal margin; the spleen was not palpable, possibly due to fluid accumulation. Bilateral leg edema was evident.
Laboratory tests on admission revealed pancytopenia; prolonged prothrombin time; hypofibrinogenemia; elevated D‐dimer level, meeting the diagnostic criteria of disseminated intravascular coagulation (DIC); severely decreased NK cell activity; and marked hyperferritinemia. Soluble IL‐2 receptor levels were elevated, a hallmark of HLH‐driven immune activation. Bone marrow examination showed hemophagocytosis, confirming excessive immune activation and increased phagocytic activity (Table 1). Contrast‐enhanced computed tomography showed hepatosplenomegaly and pronounced enhancement of the hepatic periportal region. Moderate pleural effusion was also present.
Genetic analysis using the HLH panel revealed that the patient had compound heterozygous PRF1 variants, A21V from his father and P16S from his mother (Figures 1(a) and 1(b)). Defective perforin expression in NK cells was confirmed (Figure 1(c)).
FIGURE 1Identification and functional characterization of PRF1 variants in a patient with FHL2. (a) Patient pedigree and PRF1 genotyping. Filled symbol, affected; open symbols, unaffected. (b) Details of the two missense PRF1 variants identified in the patient. (c) Flow cytometry of the perforin protein expression in NK cells from the patient and an age‐matched control stained for CD56 versus perforin. CD3^−^CD56^+^ population was analyzed.(a)(b)(c)
The patient met the HLH‐2004 diagnostic criteria based on laboratory findings at admission and was diagnosed with FHL [1]. Treatment with etoposide and liposomal steroid (dexamethasone palmitate) was initiated, and cyclosporine was added after two weeks of initial therapy, consistent with current clinical practice based on the HLH‐2004 protocol [11]. He subsequently underwent nondestructive bone marrow conditioning with thymoglobulin, fludarabine, and melphalan, followed by cord blood transplantation on Day 49 after the onset of illness. He has since survived without any FHL symptoms for over a year.
3. Discussion
In this study, we report a clinically typical case of FHL2 and provide functional characterization of two PRF1 variants, including a novel variant, A21V, and a previously reported variant in the context of heterozygous form in an adult HLH patient [12], P16S, thereby extending the genotype–phenotype correlation of PRF1‐associated FHL2.
HLH is characterized by persistent hypercytokinemia resulting from overactivated T cells and macrophages, leading to abnormal histiocyte activation and hemophagocytosis [2–4]. Upon recognition of target cells such as tumor cells and virus‐infected cells, cytotoxic T lymphocytes (CTLs) and NK cells release cytotoxic granules to target cells. Perforin, an effector molecule contained in cytotoxic granules, forms small pores in the target cell membrane to induce apoptosis. Impaired target cell killing results in persistent stimulation of CTLs and NK cells and proinflammatory cytokine overproduction that causes secondary hyperactivation of macrophages.
FHL is currently classified into five subtypes (FHL1–FHL5), all characterized by reduced cytotoxic activity due to defective perforin activity or impaired transport or release of perforin‐containing cytotoxic granules [2–4]. Although the causative genetic variants for FHL1 and the pathogenicity of FHL1 remain unidentified, the underlying genetic mechanisms for FHL2–FHL5 have been established. The current patient was diagnosed with FHL2, caused by genetic defects in the PRF1 gene encoding perforin. Therefore, perforin expression is deficient, attenuated, or functionally impaired [9].
Consistent with previous reports describing diverse PRF1 variant spectra across different ethnic groups [10], our patient exhibited a typical clinical presentation of FHL, and compound heterozygous missense variants in PRF1 were identified. The PRF1 A21V variant is currently not described in major databases, including gnomAD, ToMMo, ClinVar, HGMD, and dbSNP. However, our functional analysis showed defective PRF1 protein expression, consistent with the in silico predictions of pathogenicity using SIFT, PolyPhen‐2, and M‐CAP. Based on the patient’s typical clinical presentation and the results of functional analysis, we identified a novel PRF1 missense variant A21V as a pathogenic variant for FHL2.
The PRF1 P16S variant is currently listed in ClinVar as a variant of “Uncertain significance” and is also reported in dbSNP (rs1031606439). Although P16S has not been reported as a causative variant of AR form of FHL2, Jin et al. previously identified this variant in adult‐onset HLH patients in a heterozygous state and showed decreased PRF1 protein expression [12]. This variant was not found in the MAF, ToMMo, or gnomAD databases but was reported in dbSNP (rs1031606439). In silico analyses using SIFT, PolyPhen‐2, and M‐CAP also suggest its pathogenic potential, as evidenced by a M‐CAP score of 0.076 and classification as “Possibly Pathogenic.” Based on these findings, along with the patient’s clinical presentation and the functional analysis, we also position PRF1 P16S as a pathogenic variant causing AR form FHL2.
In conclusion, A21V is a novel PRF1 variant, and this case supports a pathogenicity of the previously reported P16S variant in autosomal recessive early‐onset FHL2 when present in trans with the A21V variant.
Author Contributions
Hiroshi Yamauchi: conceptualization, investigation, and writing–original draft.
Kazuyuki Meguro and Moeko Hino: supervision and writing–review and editing.
Taiji Nakano: validation.
Tomoko Okunushi, Hironori Sato, Yoshiharu Yamashita, and Takeshi Yamamoto: resources.
Takahiro Yasumi: methodology and validation.
Yuiko Hirata and Hirofumi Shibata: investigation.
Hiroshi Nakajima, Takahiro Aoki, and Hiromichi Hamada: writing–review and editing.
Funding
This research received no funding.
Disclosure
A preprint of this manuscript has previously been published: Hiroshi Yamauchi, et al. A case of a novel perforin gene variant in severe familial hemophagocytic lymphohistiocytosis Type 2 (FHL2) [13].
Ethics Statement
Although this study involves the off‐label use of certain drugs, their use was reviewed and approved by the Chiba University Ethics Committee (Approval number: 01‐06).
Consent
Written informed consent was obtained from the patient’s guardians after a thorough explanation of the study. Ethical considerations were prioritized, and great care was taken to ensure that the privacy of the patients was protected.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Henter J.-I. , Sieni E. , Eriksson J. et al., Diagnostic Guidelines for Familial Hemophagocytic Lymphohistiocytosis Revisited, Blood. (2024) 144, no. 22, 2308–2318, 10.1182/blood.2024025077.39046779 PMC 11619794 · doi ↗ · pubmed ↗
- 2Janka G. E. , Familial and Acquired Hemophagocytic Lymphohistiocytosis, Annual Review of Medicine. (2012) 63, no. 1, 233–246, 10.1146/annurev-med-041610-134208, 2-s 2.0-84855932987.22248322 · doi ↗ · pubmed ↗
- 3Rosado F. G. N. and Kim A. S. , Hemophagocytic Lymphohistiocytosis: An Update on Diagnosis and Pathogenesis, American Journal of Clinical Pathology. (2013) 139, no. 6, 713–727, 10.1309/ajcp 4zdkj 4icouat, 2-s 2.0-84878384497.23690113 · doi ↗ · pubmed ↗
- 4Pachlopnik Schmid J. , Côte M. , Ménager M. M. et al., Inherited Defects in Lymphocyte Cytotoxic Activity, Immunological Reviews. (2010) 235, no. 1, 10–23, 10.1111/j.0105-2896.2010.00890.x, 2-s 2.0-77951676108.20536552 · doi ↗ · pubmed ↗
- 5Jordan M. B. , Allen C. E. , Weitzman S. , Filipovich A. H. , and Mc Clain K. L. , How I Treat Hemophagocytic Lymphohistiocytosis, Blood. (2011) 118, no. 15, 4041–4052, 10.1182/blood-2011-03-278127, 2-s 2.0-80054109736.21828139 PMC 3204727 · doi ↗ · pubmed ↗
- 6Côte M. , Ménager M. M. , Burgess A. et al., Munc 18-2 Deficiency Causes Familial Hemophagocytic Lymphohistiocytosis Type 5 and Impairs Cytotoxic Granule Exocytosis in Patient NK Cells, Journal of Clinical Investigation. (2009) 119, no. 12, 3765–3773.19884660 10.1172/JCI 40732 PMC 2786810 · doi ↗ · pubmed ↗
- 7Emile J. F. , Abla O. , Fraitag S. et al., Revised Classification of Histiocytoses and Neoplasms of the Macrophage-Dendritic Cell Lineages, Blood. (2016) 127, no. 22, 2672–2681, 10.1182/blood-2016-01-690636, 2-s 2.0-84971328245.26966089 PMC 5161007 · doi ↗ · pubmed ↗
- 8Feldmann J. , Callebaut I. , Raposo G. et al., Munc 13-4 is Essential for Cytolytic Granules Fusion and is Mutated in Familial Hemophagocytic Lymphohistiocytosis (FHL 3), Cell. (2003) 115, no. 4, 461–473, 10.1016/s 0092-8674(03)00855-9, 2-s 2.0-10744224641.14622600 · doi ↗ · pubmed ↗