Prolonged Intrahepatic Cholestasis After Acute Hepatitis E Infection: A Case Series and Genetic Analysis
Montserrat Fraga, Sophie Kasmi, Susanne N. Weber, Roman Liebe, Christine Sempoux, Ali Saadat, Jacques Fellay, Silke Kenngott‐Kelber, Vincent Zimmer, Frank Lammert, Marcin Krawczyk, Christoph Jüngst

TL;DR
This study explores how genetic factors may contribute to prolonged liver issues after Hepatitis E infection in some patients.
Contribution
The study identifies specific genetic variants potentially linked to prolonged cholestasis following HEV infection in immunocompetent individuals.
Findings
Two patients had potentially pathogenic genetic variants (ATP8B1 p.N45T and MYO5B p.K429R) associated with prolonged cholestasis.
Common ABCB11 variants were found in all patients, possibly contributing to cholestatic symptoms.
The MYO5B p.K429R variant was absent in asymptomatic HEV-infected controls.
Abstract
Hepatitis E virus (HEV) is a leading cause of acute viral hepatitis worldwide. Whereas HEV infection is typically self‐limiting, rare cases of prolonged cholestasis have been reported. The underlying mechanisms remain unclear, though host genetic variation may contribute. This study aimed to investigate the role of genetic predisposition in HEV‐induced prolonged cholestasis by analysing variants in genes associated with hepatocanalicular transport. We performed a retrospective review of medical records from three university centres in Switzerland and Germany and identified five immunocompetent patients with prolonged cholestasis following acute HEV infections. Genetic analysis using next‐generation sequencing included a panel of five genes involved in cholestatic liver diseases (ATP8B1, ABCB11, ABCB4, ABCC2 and MYO5B). Variant frequencies were evaluated using population reference…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
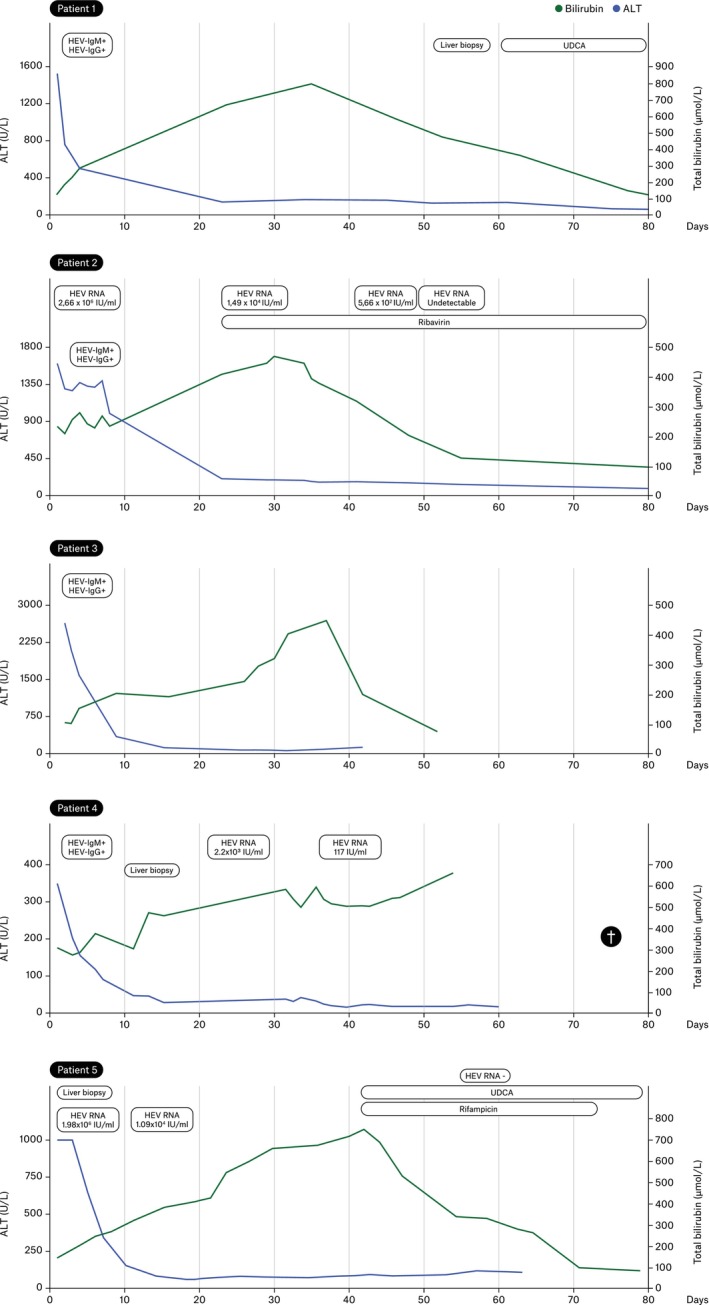 FIGURE 1
FIGURE 1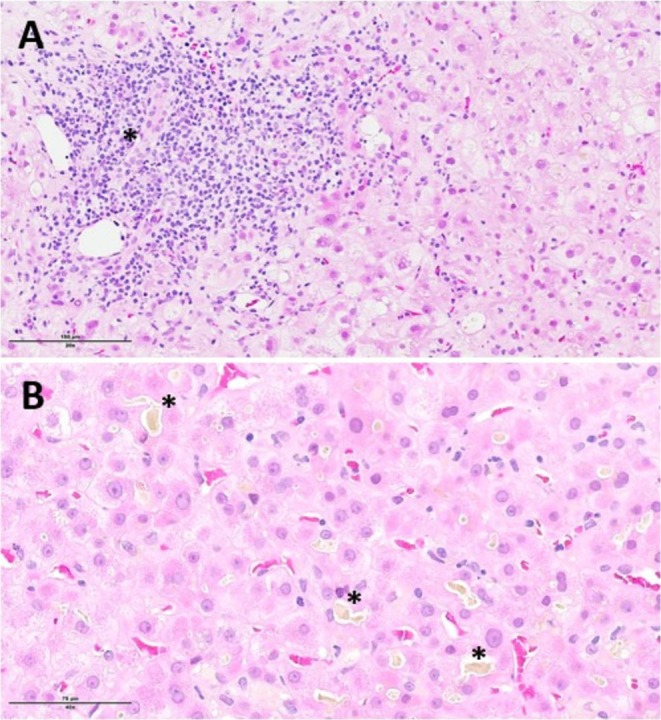 FIGURE 2
FIGURE 2| Patient | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Age (years) | 59 | 72 | 36 | 83 | 50 |
| Sex | Male | Male | Male | Male | Male |
| Ethnicity | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian |
| Serology (IgM/IgG) | IgM+/IgG+ | IgM+/IgG+ | IgM+/IgG+ | IgM+/IgG+ | IgM+/IgG+ |
| HEV RNA (IU/ml) | Negative (d44 of jaundice) | 2.6 × 106 | Negative (d28 of jaundice) | 2.2 × 103 | 1.9 × 106 |
| ALT peak (U/L) | 1514 | 1606 | 2646 | 359 | > 1000 |
| ALP peak (U/L) | NA | 208 | 157 | 188 | 304 |
| Total bilirubin peak (μmol/L) | 795 | 469 | 452 | 578 | 736 |
| GGT peak (U/L) | NA | 433 | 205 | 320 | 389 |
| Jaundice duration (days) | 83 | 88 | > 52 | > 60 | > 77 |
| Histology | Canalicular cholestasis, no hepatitis, no cholangitis | — | — | Severe cholestatic hepatitis | Canalicular cholestasis |
| Treatment | UDCA | Ribavirin | — | — | UDCA, Rifampicin |
| Clinical outcome | Cholestasis resolution | Cholestasis resolution | Cholestasis resolution | Death | Cholestasis resolution |
| Variant | Location and nucleotide alteration | Predicted effect | P1 | P2 | P3 | P4 | P5 |
|---|---|---|---|---|---|---|---|
|
| |||||||
| rs146599962 |
|
| Heterozygous | ||||
| rs319438 | c. 696 T > C | p.D232D | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous |
| rs319439 | c.698 + 20C > T | Heterozygous | |||||
| rs319443 | c.811A > C | p.R271R | Homozygous | Homozygous | Homozygous | Homozygous | Homozygous |
| c.2098‐8C > A | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |||
| c.2098‐4C > A | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |||
| rs12968116 | c.2855G > A | p.R952Q | Heterozygous | ||||
| rs222581 | c.3454G > A | Homozygous | Homozygous | Homozygous | Homozygous | ||
|
| |||||||
|
| |||||||
| rs4148776 | c.99‐18 T > C | Heterozygous | |||||
| rs11568363 | c.389 + 8G > A | Heterozygous | |||||
| c.909–17,909–15 | Heterozygous | ||||||
| rs2287618 | c.909–15A > G | Heterozygous | Heterozygous | Heterozygous | |||
| rs7563233 | c.957A > G | p.G319G | Heterozygous | ||||
| rs2287622 | c.1331C > T | p.A444V | Heterozygous | Heterozygous | Heterozygous | ||
| rs11568364 | c.2029A > G | p.M677V | Heterozygous | ||||
| rs853772 | c.2179‐17C > A | Homozygous | Heterozygous | Homozygous | Heterozygous | Homozygous | |
| rs853789 | c.2344‐17 T > C | Homozygous | Heterozygous | Homozygous | Heterozygous | Homozygous | |
| rs497692 | c.3084A > G | p.A1028A | Homozygous | Heterozygous | Homozygous | Heterozygous | Homozygous |
| c.3882G > T | p.V1294V | Heterozygous | |||||
|
| NA | ||||||
| rs2302387 | c.175C > T | Heterozygous | Heterozygous | Heterozygous | |||
| rs1202283 | c.504C > T | p.L59L | Heterozygous | Heterozygous | Heterozygous | Homozygous | |
| rs2109505 | c.711A > T | p.N168N | Heterozygous | Heterozygous | |||
| rs2230028 | c.1954A > G | p.I237I | Heterozygous | ||||
| rs31668 | c.2211 + 16C > T | p.R652G | Heterozygous | Homozygous | Homozygous | Heterozygous | |
| rs31653 | c.3508‐16 T > C | Heterozygous | Heterozygous | Homozygous | Heterozygous | ||
|
| NA | ||||||
| rs1815930 | c.376A > G | p.T126A | Homozygous | Homozygous | Homozygous | Homozygous | |
| rs17659179 | c.921G > T | p.K307K | Heterozygous | ||||
|
|
| Heterozygous | |||||
| rs17715416 | c.1545 + 11 T > C | Homozygous | |||||
| c.1586A > G | Heterozygous | ||||||
| rs2298628 | c.2049G > A | p.V683V | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |
| rs2298624 | c.2735G > A | p.R919H | Heterozygous | Heterozygous | |||
| rs200219597 | c.3163_3165dup | p.L1056dup | Heterozygous | Homozygous | Heterozygous | ||
| rs2276176 | c.3276 + 11 T > C | Heterozygous | Heterozygous | ||||
| rs3826579 | c.3591C > T | p.Y1197Y | Heterozygous | Heterozygous | |||
| rs488890 | c.4315 + 5G > C | Homozygous | Heterozygous | ||||
| rs112417235 | c.5062A > G | p.M1688V | Heterozygous | ||||
|
| NA | ||||||
| rs927344 | c.116A > T | p.Y39F | Homozygous | Homozygous | Homozygous | Homozygous | |
| rs17222561 | c.1483A > G | p.K495E | Heterozygous | ||||
| rs3740066 | c.3972C > T | p.I1324I | Heterozygous | Homozygous | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHepatitis Viruses Studies and Epidemiology · Liver Diseases and Immunity · Liver Disease and Transplantation
Introduction
1
Hepatitis E virus (HEV), a small, non‐enveloped, positive‐sense single‐stranded RNA virus, is a leading cause of jaundice and hepatitis worldwide [1]. Approximately 950 million people, or about 12.5% of the global population, have been infected with HEV [2].
There are two distinct clinical and demographic scenarios. HEV genotypes 1 and 2, found in developing countries, infect only humans and spread via the faecal‐oral route. These genotypes cause hepatitis outbreaks in areas with poor sanitation and high mortality rates, particularly in pregnant women [1, 3]. In industrialised countries, pigs are the primary reservoir for HEV genotypes 3 and 4, and zoonotic transmission to humans typically occurs through the consumption of undercooked or raw meat [1, 3].
In immunocompetent individuals, acute autochthonous HEV infection is usually asymptomatic and self‐limiting [1, 3]. Extrahepatic manifestations mainly include neurological conditions, such as Guillain‐Barré and Parsonage‐Turner syndromes [1, 4]. In immunosuppressed individuals, chronic HEV infection may progress from chronic hepatitis to cirrhosis [1, 5]. Finally, rare cases of benign recurrent intrahepatic cholestasis (BRIC)‐like episodes with prolonged jaundice have been reported after acute HEV infection [6, 7, 8, 9].
The mechanisms underlying this broad range of clinical manifestations remain unclear, although host genetics is being increasingly discussed. A recent study [10] identified genetic variants related to the type I interferon response in patients with acute symptomatic and severe hepatitis after autochthonous HEV infection. Furthermore, two recent case reports suggested a genetic predisposition of hepatocanalicular transporter genes to developing a BRIC‐like episode after acute HEV infection [9, 11].
Therefore, we hypothesised that host genetic factors might play a role in influencing HEV‐induced prolonged cholestasis and explored the genetic contributions in otherwise healthy individuals who experienced prolonged cholestasis triggered by HEV.
Material and Methods
2
Study Population and Design
2.1
We conducted a retrospective review of the medical records of patients with prolonged cholestatic jaundice following a confirmed acute HEV infection at three university centres in Switzerland (Lausanne, Zürich) and Germany (Homburg). The electronic records and archives were systematically examined. Informed consent was obtained for all patients. The local ethics committee approved this study (Ärztekammer des Saarlandes, #271/11; CER‐VD protocol #2020–00197).
The inclusion criteria were [1] confirmed HEV infection, verified by positive HEV RNA in plasma and/or stool via polymerase chain reaction (PCR) testing, or by detection of anti‐HEV immunoglobulin M (anti‐HEV IgM); and [2] prolonged cholestatic jaundice following acute HEV infection, defined as total bilirubin levels exceeding 50 μmol/L for more than 30 days.
The exclusion criteria were as follows [1] patients with pre‐existing alcohol use disorder, chronic liver disease, advanced fibrosis/cirrhosis or immunosuppression [2] patients with alternative potential causes of cholestasis, including viral hepatitis other than HEV infection, autoimmune diseases, drugs or herbal supplements and biliary obstruction (systematically ruled out through abdominal ultrasound and/or magnetic resonance cholangiography).
Baseline Evaluation
2.2
The demographic data included age, sex and ethnicity. Clinical data encompassed relevant comorbidities, history of chronic liver disease, alcohol use disorder, ongoing treatments at presentation and any treatment for acute hepatitis E. Laboratory data included liver function tests, total bilirubin, serum albumin, coagulation parameters and creatinine levels. Virological data included anti‐HEV IgG and IgM serology, quantitative HEV RT‐PCR in plasma and/or stool samples. Pathological data included expert liver biopsy assessments when available. Genetic data consisted of next‐generation sequencing (NGS) for five genes associated with cholestatic liver conditions.
Next Generation‐Sequencing and Bioinformatic Analysis
2.3
Genomic DNA from peripheral EDTA blood was isolated using standard procedures (QiAamp DNA extraction; Qiagen, Hilden Germany). Library preparation for NGS was performed using Illumina DNA prep with enrichment (Illumina, San Diego, USA). Primers for the distinct genes (ATP8B1, ABCB11, ABCB4, ABCC2 and MYO5B) were designed as custom panels in cooperation with Illumina.
Direct paired‐end NGS sequencing (2 × 150 bp) was performed on the Illumina MiniSeq platform, and the results were analysed using JSI SeqNext software (JSI, Ettenheim, Germany) at the Saarland University Medical Center, Germany. In patient 5 paired‐end NGS sequencing of solely ATP8B1 and ABCB11 was performed as part of the routine diagnostic procedure by the Institute of Medical Genetics at the University of Zurich, Switzerland. To gain further insight into the potential importance of the detected variants, we assessed their frequencies in the Genome Aggregation Database (gnomAD) (gnomad.broadinstitute.org).
Comparison Cohort
2.4
As a comparison cohort, we included asymptomatic HEV‐infected blood donors from a previously published Swiss multicentre genomic study [10]. Asymptomatic blood donors were identified through routine nationwide blood donation screening performed in Switzerland between January 2021 and October 2022, during which systematic HEV testing was conducted in accordance with national blood safety regulations. Acute HEV infection was confirmed by quantitative polymerase chain reaction (PCR). All individuals were adults (≥ 18 years) and underwent standardised clinical assessment using a structured questionnaire to ensure the absence of symptoms related to HEV infection. Only strictly asymptomatic donors were included. All these asymptomatic blood donors included in the comparison cohort had undergone whole‐genome sequencing (WGS) as part of the previously published Swiss multicentre genomic study.
Results
3
Study Participants
3.1
Five patients with acute hepatitis E‐induced prolonged cholestasis between May 2013 and February 2022 were retrospectively identified. Notably, patient 1 was already reported [9]. Extensive diagnostic workups, including imaging, ruled out biliary obstruction and alternative causes of jaundice.
All patients were men and of Caucasian origin with a median age of 59 years (range 36–83 years). Table 1 summarises the patients' characteristics and Figure 1 illustrates the clinical course of the five patients.
Clinical course of the five identified patients. UDCA, ursodeoxycholic acid; †, death.
The median alkaline phosphatase peak value was 198 U/L (range 157–304 U/L). The median gamma‐glutamyltranspeptidase (GGT) peak activity was 354 U/L (range 205–433 U/L). In all five patients, the GGT levels were low during the prolonged cholestasis. The median peak total bilirubin concentration was 578 μmol/L (range 452–795 μmol/L). The median duration of total bilirubin levels > 50 μmol/L was 77 days (range 52–88 days). In patient 4, serum bile acids measured on day 32 after presentation were elevated at 56 μmol/L (reference value ≤ 10 μmol/L).
Three patients (patients 1, 4 and 5) underwent a liver biopsy. In patients 1 and 5, the biopsy showed canalicular cholestasis without signs of hepatitis or chronic cholangitis. A liver biopsy of patient 4 was performed 10 days after his initial presentation, demonstrating features of cholestatic hepatitis without fibrosis (Figure 2). One patient was treated with ursodeoxycholic acid (UDCA), and one patient was treated with ribavirin for 4 weeks until HEV RNA was undetectable in the blood and stool. A third patient was initially treated with UDCA without any impact on bilirubin levels and itch, and after additional treatment with rifampicin (150 mg/day), there was a prompt continuous improvement in bilirubin levels and itching. Two patients did not receive any specific drug treatment. Complete resolution of cholestasis was observed in four patients. Patient 4, an 83‐year‐old male, developed intractable pruritus with an unfavourable course of cholestasis. The patient's condition was further complicated in the late disease course by bacterial pneumonia, leading to death.
Results of liver biopsy in patient 4. A. Portal tracts: Diffuse inflammatory infiltration, mostly lymphocytic, with florid interface activity and no bile duct lesion () or significant ductular reaction. B. Lobules: Numerous bile thrombi in the canaliculi ().
Comparison Cohort
3.2
The comparison cohort consisted of 16 asymptomatic blood donors with confirmed acute HEV infection [10]. Twelve of the 16 participants (75.0%) were male, with a median age of 56 years (range, 24–74 years). All individuals were of Caucasian origin. None had a history of chronic liver disease, and none were receiving immunosuppressive therapy at the time of diagnosis. No participant reported recent travel to regions endemic for HEV genotypes 1 or 2, supporting autochthonous HEV infection consistent with genotype 3 circulation in Switzerland. All individuals were asymptomatic at the time of HEV diagnosis, as documented by a standardised questionnaire specifically designed to exclude clinical manifestations of HEV infection. Quantitative HEV RNA PCR was performed in all participants, with a median viral load of 3.1 log_10_ IU/mL (range, 1.1 – 5.3 log_10_ IU/mL).
Genetic Analysis
3.3
Genetic analysis revealed multiple variants in the five genes analysed (Table 2). Based on gnomAD frequency (AF) in Europeans (non‐Finnish) and previously published findings, the following variants, observed in a heterozygous state, were considered potentially pathogenic: the rare missense variant ATP8B1 p.N45T (gnomAD AF = 0.0055), which was detected in patient 1 [12, 13], and the ultra‐rare variant MYO5B p.K429R (gnomAD AF = 0.0000017), which was observed in patient 2.
All other variants were considered benign or likely benign, although several have been described as minor contributors towards impaired bile secretion [14, 15].
WGS was performed in all 16 asymptomatic HEV‐infected blood donors as part of a previously published original research study [10]. Targeted analysis of the two variants of interest revealed that the MYO5B p.K429R variant was absent in all asymptomatic HEV‐infected blood donors, whereas the ATP8B1 p.N45T variant was detected in only one control individual and exclusively in the heterozygous state.
Discussion
4
Although acute HEV infection is typically asymptomatic and self‐limiting, it can occasionally result in severe hepatitis and jaundice. It has been shown that host genetics may influence the clinical presentation of HEV infections [10]. Based on this background, we hypothesised that functional variants within hepatocanalicular bile salts or phospholipid transporters might contribute to severe and prolonged forms of HEV‐induced cholestasis [9].
Homozygous functional variants of genes involved in biliary lipid transport may cause severe forms of hereditary cholestasis, collectively termed progressive familial intrahepatic cholestasis (PFIC). Heterozygous variants of ATP8B1 and ABCB11 have been linked to intermittent prolonged cholestatic episodes, also known as benign recurrent intrahepatic cholestasis (BRIC) types 1 and 2, respectively [16]. Cholestasis with jaundice typically lasts several weeks to months in patients with BRIC, and GGT levels are typically low. Various trigger factors have been described such as drugs, hormones, surgery and viral infections especially in the upper respiratory tract [17]. Cholestatic phenotypes arise from an interplay between genetic predisposition and environmental factors, whereby carriers of risk variants exhibit cholestasis only when additional triggers compromise transporter expression or function, while remaining phenotypically unaffected in their absence.
We analysed five patients who presented with prolonged icteric cholestasis following acute HEV infection to investigate potential variants in genes associated with hepatocellular cholestasis. As a comparison cohort, we included asymptomatic blood donors with documented acute HEV infection who had undergone WGS in a previous study.
Genetic analysis of patients with HEV‐induced cholestatic hepatitis revealed a complex picture of multiple variants (Table 1), two of which deserve further attention.
To place our genetic findings in an appropriate population context, we assessed the frequency of all detected variants using the Genome Aggregation Database (gnomAD) to distinguish common background variation from potentially relevant rare alleles. Two heterozygous variants were retained based on their low population frequency and prior published data: ATP8B1 p.N45T, a rare missense variant (gnomAD European non‐Finnish AF = 0.0055), and MYO5B p.K429R, an ultra‐rare variant (gnomAD AF = 0.0000017). These frequencies indeed indicate that both variants are uncommon in the general population, with MYO5B p.K429R being particularly rare.
In patient 1, we identified the heterozygous variant ATP8B1 c.134A > C, resulting in an amino acid substitution of asparagine with threonine (p.N45T). ATP8B1 acts as a flippase at the hepatocanalicular membrane and translocates phospholipids from the external to the cytoplasmic membrane leaflet and thereby maintains the canalicular membrane asymmetry. A disrupted membrane symmetry leads to impaired canalicular protein function, including the bile salt export pump (BESP), which is encoded by the ABCB11 gene and represents the primary transporter of bile salts from the hepatocyte into bile, thereby contributing to cholestasis [18, 19]. This ATP8B1 variant was initially described in a woman with intrahepatic cholestasis of pregnancy (ICP) [13]. Dixon et al. [12] identified six patients carrying the ATP8B1 p.N45T mutation in a larger ICP cohort, in which the variant was observed at approximately twice the frequency reported in the general population. Importantly, the global minor allele frequency of this variant in gnomAD population reference databases is low and approximately 0.5%. This variant was also identified in the heterozygous form in a patient with BRIC [20].
In a second patient, we detected an ultra‐rare heterozygous variant in the MYO5B gene, resulting in the substitution of lysine with arginine (p.K429R). Notably, the global population prevalence of this variant is extremely low, estimated at 0.00017% in the gnomAD reference dataset. MYO5B encodes a protein involved in cell polarisation and the intracellular trafficking of transport proteins, such as BSEP to the canalicular membrane. Severe MYO5B mutations result in microvillus inclusion disease, a pronounced intestinal phenotype, whereas isolated cholestatic liver diseases such as chronic low GGT liver disease or recurrent episodes of cholestasis have also been reported [18, 21, 22]. The MYO5B variant p.K429R is very rare according to gnomAD (present in only two out of 1,180,010 alleles). The variant was localised in a highly conserved region of the gene, suggesting that it is functionally relevant. Furthermore, it has been shown that liver phenotypes result from relatively mild MYO5B variants rather than biallelic nonsense or frameshift mutations and can also occur in heterozygous carriers [22].
The rarity of the two identified variants supports a genetic susceptibility model for prolonged cholestatic hepatitis following acute HEV infection. Consistent with this, comparison with a WGS‐characterised asymptomatic HEV‐infected blood donor cohort showed absence of the ultra‐rare MYO5B p.K429R variant and only a single heterozygous carrier of the ATP8B1 p.N45T variant. Despite limited sample size, these findings suggest that such variants are uncommon in uncomplicated HEV infection and may contribute to susceptibility to prolonged cholestasis.
Interestingly, a comparable susceptibility framework has been described in cholestatic drug‐induced liver injury (DILI). Although the specific variants identified in our cohort (ATP8B1 p.N45T and MYO5B p.K429R) have not been reported in DILI, variants in other cholestasis‐related genes from our panel (particularly ABCB11 and ABCB4) have been linked to cholestatic DILI. These observations suggest overlap at the gene and pathway level between cholestatic DILI and HEV‐associated cholestasis, despite differences at the individual variant level [23, 24].
Overall, we consider that both variants, ATP8B1 p.N45T and MYO5B p.K429R, might have contributed to the observed phenotype, with the limitation that functional analyses are lacking. Several of the common variants detected in the ABCB11 gene might also add to predisposition to cholestasis in these patients. The ABCB11 c.3084A > G synonymous variant was found in all patients, either in the heterozygous or homozygous states. This risk variant has been linked to the development of preoperative jaundice and decreased protein and mRNA expression in patients with intrahepatic gallstones [15]. The ABCB11 c.909–15A > G variant has been associated with a reduced response to ursodeoxycholic acid in patients with primary biliary cholangitis and was observed in three of the patients in our study [14].
In conclusion, NGS analysis of the total coding regions for the three most frequent PFIC‐associated genes ATP8B1, ABCB11 and ABCB4 (PFIC1‐3), as well as MYO5B (PFIC6) and ABCC2 revealed potentially pathogenic variants in two patients, and several predisposing common ABCB11 variants in all patients. The causative genes for the very rare subtypes PFIC4 (TJP2) and PFIC5 (NR1H4) were not analysed.
Our findings align with those of previous reports that have highlighted the influence of host genetics in modulating cholestatic responses to viral infections. A similar observation was reported in a case of hepatitis A virus (HAV)‐induced cholestasis [25], a virus that shares important clinical commonalities with HEV [26]. Our findings are further supported by a recent case report by Drexler et al., who described two immunocompetent patients with prolonged intrahepatic cholestasis following acute HEV infection, in the absence of pre‐existing liver disease and of other identifiable causes [11]. Similar to our cohort, both patients exhibited low GGT levels, marked hyperbilirubinemia and histological features consistent with canalicular cholestasis. Next‐generation sequencing revealed heterozygous variants in ATP8B1 and other hepatobiliary transporter genes, including ABCB4, ABCB11 and MYO5B, supporting the hypothesis that host genetic background may modulate the severity of cholestatic responses to HEV. In both studies, the cholestatic phenotype occurred in patients without prior liver disease, reinforcing the concept that HEV may act as a precipitating factor in genetically predisposed but otherwise healthy individuals.
Genetic assessment of patients experiencing prolonged cholestasis following acute viral hepatitis could enhance the personalised infection management in such cases. Targeted interventions, such as the early initiation of drugs that have been shown to interact with the enterohepatic circulation of bile acids such as rifampicin or ileal bile acid transporter (IBAT) inhibitors, could be considered [27].
While the cohort size was limited, our study presents the most extensive series to date of prolonged cholestasis after acute HEV infection, including genetic data. Our experience indicates that the severity of HEV infection may be influenced by host genetics as evidenced by the prolonged cholestatic jaundice in genetically predisposed individuals. These findings underscore the value of integrating genetic analyses to enhance our understanding of the complex pathogenesis, which has also been emphasised in recent guidelines on genetic cholestatic liver disease [28].
Author Contributions
M.F., and C.J., selected the patients. All authors contributed to the collection of patients' data and analysis. M.F., C.J., and S.K., wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1N. Kamar , J. Izopet , N. Pavio , et al., “Hepatitis E Virus Infection,” Nature Reviews Disease Primers 3 (2017): 17086.10.1038/nrdp.2017.8629154369 · doi ↗ · pubmed ↗
- 2P. Li , J. Liu , Y. Li , et al., “The Global Epidemiology of Hepatitis E Virus Infection: A Systematic Review and Meta‐Analysis,” Liver International 40, no. 7 (2020): 1516–1528.32281721 10.1111/liv.14468 PMC 7384095 · doi ↗ · pubmed ↗
- 3N. Kamar , R. Bendall , F. Legrand‐Abravanel , et al., “Hepatitis E,” Lancet 379, no. 9835 (2012): 2477–2488.22549046 10.1016/S 0140-6736(11)61849-7 · doi ↗ · pubmed ↗
- 4H. R. Dalton , R. P. Bendall , M. Rashid , et al., “Host Risk Factors and Autochthonous Hepatitis E Infection,” European Journal of Gastroenterology & Hepatology 23, no. 12 (2011): 1200–1205.21941192 10.1097/MEG.0b 013e 32834 ca 4da · doi ↗ · pubmed ↗
- 5S. J. Wallace , R. Swann , M. Donnelly , et al., “Mortality and Morbidity of Locally Acquired Hepatitis E in the National Scottish Cohort: A Multicentre Retrospective Study,” Alimentary Pharmacology & Therapeutics 51, no. 10 (2020): 974–986.32285976 10.1111/apt.15704 · doi ↗ · pubmed ↗
- 6S. B. Ji , S. S. Lee , H. C. Jung , et al., “A Korean Patient With Guillain‐Barré Syndrome Following Acute Hepatitis E Whose Cholestasis Resolved With Steroid Therapy,” Clinical and Molecular Hepatology 22, no. 3 (2016): 396–399.27572076 10.3350/cmh.2015.0039 PMC 5066380 · doi ↗ · pubmed ↗
- 7L. Mechnik , N. Bergman , M. Attali , et al., “Acute Hepatitis E Virus Infection Presenting as a Prolonged Cholestatic Jaundice,” Journal of Clinical Gastroenterology 33, no. 5 (2001): 421–422.11606863 10.1097/00004836-200111000-00017 · doi ↗ · pubmed ↗
- 8R. Moucari , J. Bernuau , E. Nicand , et al., “Acute Hepatitis E With Severe Jaundice: Report of Three Cases,” European Journal of Gastroenterology & Hepatology 19, no. 11 (2007): 1012–1015.18049173 10.1097/MEG.0b 013e 328209414 d · doi ↗ · pubmed ↗