Photoactivation in Chemotherapeutic Compounds for Cancer Treatment: Opportunities Beyond Photodynamic Therapy
Simon Ngigi Mbugua, Eunice Adhiambo Nyawade

TL;DR
This paper reviews photoactivatable anticancer drugs that use light to control drug activity, offering benefits like reduced side effects and better targeting of tumors.
Contribution
The paper provides a comprehensive review of photoactivatable anticancer compounds and their advantages over traditional chemotherapy.
Findings
Photoactivatable drugs allow precise control of drug activity using light, reducing off-target effects.
These compounds can overcome drug resistance and enable in situ monitoring of drug activation.
The paper explores synthesis methods and effectiveness of various photoresponsive metal complexes.
Abstract
Photoactivation is the stimulation or regulation of a chemical or a chemical process by utilizing light of specific wavelength that corresponds to an absorbance optimum of the agent being used and can penetrate into tissues. In cancer therapy, photoactivatable drugs utilize this phenomenon by allowing the temporal and spatial regulation of their cytotoxicity using irradiation. Therefore, in order to reduce the adverse effects of platinum medications, photoactivatable anticancer pharmaceuticals, which might be site‐activated in the tumour region, are a viable option. This paper summarizes different types of photoactivatable anticancer compounds that would produce an active version of a drug by the process of photouncaging. The mode of photoactivation and rationale for drug design are summarized. The effects of typical complexes on cellular pathways, photocytotoxicity and dark…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 SCHEME 1
SCHEME 1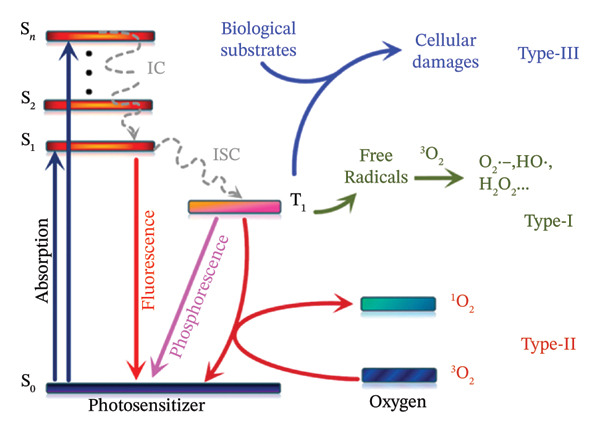 FIGURE 1
FIGURE 1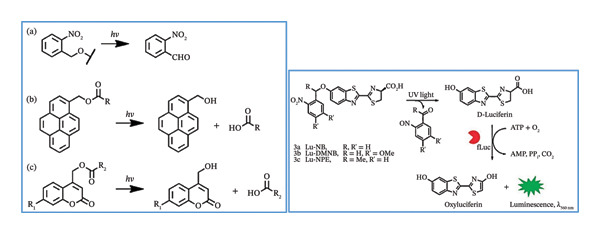 SCHEME 2
SCHEME 2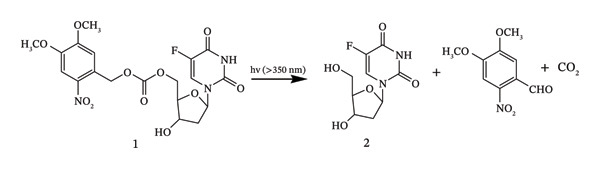 SCHEME 3
SCHEME 3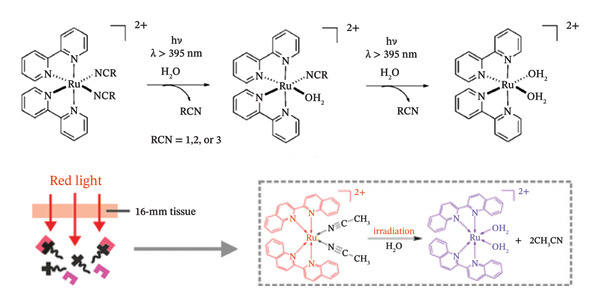 FIGURE 3
FIGURE 3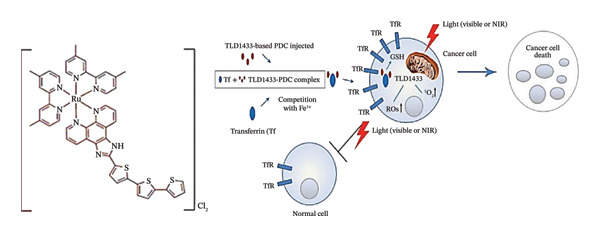 FIGURE 4
FIGURE 4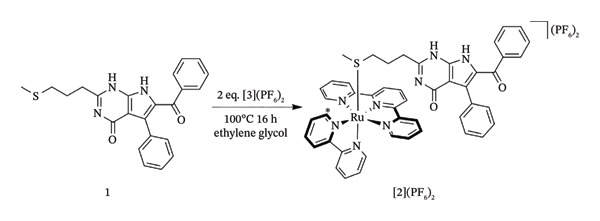 SCHEME 4
SCHEME 4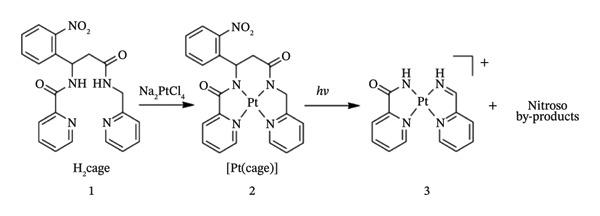 FIGURE 6
FIGURE 6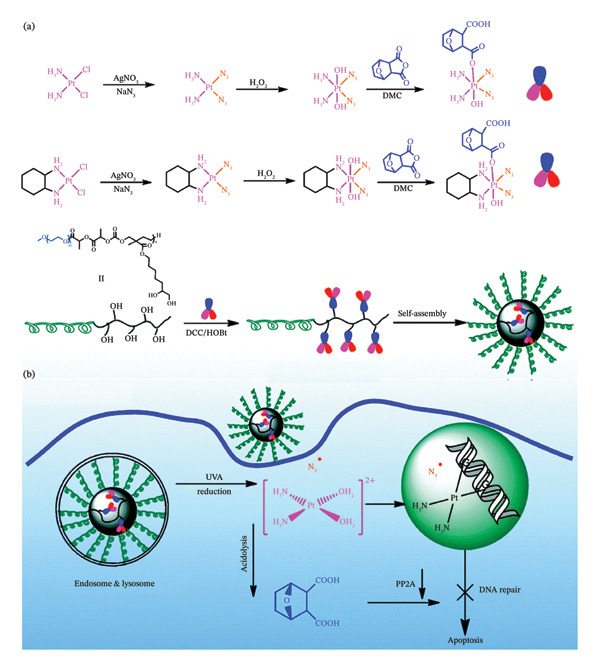 FIGURE 7
FIGURE 7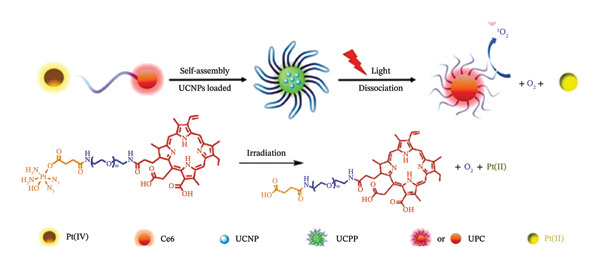 FIGURE 9
FIGURE 9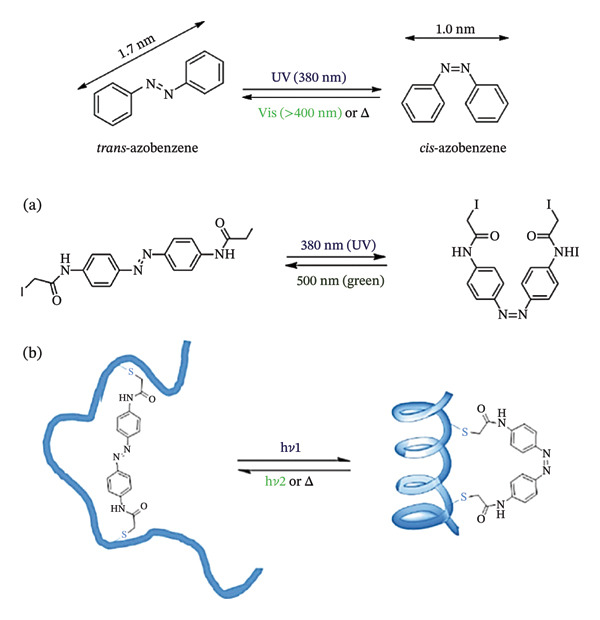 SCHEME 6
SCHEME 6| Factor | Ru(II) complexes | Pt complexes | References |
|---|---|---|---|
| Photoresponsiveness | Strong (visible/NIR light possible); MLCT transitions ideal | Moderate (mostly UV, some visible‐responsive Pt(IV) systems) | [ |
| Photoactivation mechanism | Ligand dissociation, ROS generation or prodrug release | Photoreduction (Pt(IV ⟶ II)), ligand photodissociation | [ |
| Activation wavelength | Tunable (300–770 nm; some NIR designs) | Typically UV (320–400 nm); NIR‐active Pt complexes still emerging | [ |
| Excited‐state lifetime | Long‐lived (microseconds); enhances photocleavage efficiency | Shorter lifetimes; less efficient photochemistry unless engineered | [ |
| Cytotoxicity (dark) | Generally low; stable in biological conditions | Pt(IV) prodrugs have low dark toxicity; Pt(II) photocaged forms may be more toxic | [ |
| Cytotoxicity (light) | High cytotoxicity postactivation; DNA/RNA damage, ROS | High; photoactivated Pt(II) forms form DNA crosslinks or adducts | [ |
| Selectivity and control | High spatiotemporal precision; often used with targeting ligands | Reasonable control with prodrug design; UV limits precision in tissues | [ |
| Biocompatibility | High; good aqueous solubility and tunable pharmacokinetics | Moderate to good; Pt(IV) prodrugs designed for improved compatibility | [ |
| Theranostic capability | Strong luminescent for imaging and therapy | Limited nonluminescent unless modified | [ |
| Resistance profile | Less prone to cisplatin‐type resistance mechanisms | Pt(IV) can evade some resistance; Pt(II) complexes still face efflux, detox issues | [ |
| Delivery enhancements | Often integrated into liposomes, nanoparticles, conjugates | Similar strategies used; lipophilic cages can aid delivery | [ |
| Structural change | Effect on photoactivation | Effect on cytotoxicity | Effect on targeting |
|---|---|---|---|
| Presence of electron‐donating groups | Reduction in activation energy | Increase or decrease (depends on target) | No direct effect |
| Steric effects around metal centre | Increases ligand photodissociation by destabilizing the ground state | Enhances cytotoxicity (if release is cytotoxic) | May affect delivery |
| Presence of targeting moieties | No effect | No effect | Enhancement of targeting specificity |
| Use photolabile axial ligands in Pt(IV) complexes | Greatly increases rates of photoreduction | Raises cytotoxicity under light | May improve binding to DNA nucleobases |
| Presence of intercalating ligands | Increases DNA interactions | Enhances dark toxicity | Improves nuclear targeting |
| Structural modification | Electronic effect |
|---|---|
| Electron‐donating groups on ligand groups | Lowers MLCT transition energy resulting to a red‐shift absorption making the complex more responsive to visible light [ |
| π‐extended ligands | Enhance absorption cross‐section and intersystem crossing leading to longer‐lived excited states. This in turn may result into higher activation yields [ |
| Sterically bulky ligands at the metal centre | Destabilizes the ground state leading to easier ligand dissociation during irradiation [ |
| Chelation mode (monodentate vs. polydentate ligands) | Monodentate ligands are more photolabile and easier to release compared to polydentate chelates [ |
| Use of with photolabile ligands (e.g., nitriles, thiocarbonyls) | Increases sensitivity to light leading to efficient ligand dissociation [ |
| Compound | Mechanism | Stage | References |
|---|---|---|---|
| TLD‐1433 | ROS production, PDT | Clinical trials (bladder cancer) | [ |
| Pt(IV)‐DACH prodrugs | Pt(II) release, DNA binding | Preclinical | [ |
| [Ru(bpy)2(L)]2+ with caged HDAC inhibitors | Enzyme inhibition | Lab studies | [ |
| Pt(IV)‐diazido complexes | ROS + Pt(II) generation | Preclinical | [ |
| [Ru(bpy)2(L‐drug)]2+ photocages | A cytotoxic small molecule (such as an analogue of nicotinamide or a NAMPT inhibitor) is released when exposed to light; this results in cytotoxicity. | Preclinical – cell and animal studies only. No human trials to date. | [ |
| Ru‐caged microtubule‐inhibitor (red‐light activatable) | Microtubule polymerization inhibitor induced by light; shown in vitro and in vivo models | Preclinical (recent reports, in vivo tumour models). | [ |
| Ru‐STF31/Ru‐linked metabolic inhibitors | NAD+ depletion and metabolic disruption in cancer cells are caused by the release of metabolic/targeted inhibitors (such as STF31 derivatives). | Preclinical (cell experiments reported; early animal data in some studies). | [ |
| Ru(II) photocages for enzyme inhibitors/CYP inhibitors (Havrylyuk et al.) | Accurate regulation of enzyme inhibitors (proof‐of‐concept photocages that, when exposed to light, release active inhibitors) | Preclinical/chemical‐biology tools | [ |
| Dual‐action Ru complexes (PACT + PDT) (recent designs) | To increase effectiveness and get over hypoxic restrictions, combine a photo‐released medication with photosensitization. | Preclinical (concept and animal data reported) | [ |
| Ru‐DOX/polymeric Ru precursors for DOX photorelease | Chemotherapeutic release driven by light or photo‐enhanced antimicrobial activity (proof of concept) | Preclinical | [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNanoplatforms for cancer theranostics · Photodynamic Therapy Research Studies · Photochromic and Fluorescence Chemistry
1. Introduction
The inability of anticancer metallodrugs to distinguish between normal and malignant cells is a significant disadvantage to a certain extent leading to systemic toxicity. On the other hand, photoresponsive metal complexes are a potential class of anticancer treatments with enhanced selectivity [1–3]. Light can regulate metal complexes’ toxicity, making them minimally intrusive and spatially selective, all under a remotely managed approach. One of the main objectives of drug delivery technology is to be able to manage medication dosage in terms of amount, location and time. Better control enhances therapeutic efficacy while reducing negative effects. Stimulated delivery systems have been presented as systems that respond to an external stimulus, such as temperature, pH, applied magnetics, electrical or light fields, ultrasound, light or enzymatic activity [4, 5].
Transition metal complexes have proven effective in antitumour phototherapies during the past few decades. In several domains, which include photoinduced ligand exchange or release, diverse excited state activity and adaptable biochemical features, they have demonstrated encouraging characteristics [6, 7]. Photoresponsive metallopolymer nanoparticles (MPNs), for example, exhibit improved hydrosolubility, prolonged blood circulation and elevated tumour‐specific retention when integrated into polymeric systems and nanostructures [8]. This results in a significant improvement in tumour treatment efficacy when in comparison with low‐molecule‐weight metallic complexes [8].
The current state of pharmaceutical treatments employed in cancer chemotherapy is significantly impacted by their harmful side effects and low selectivity. The majority of pharmacological molecules now on the market are not selective to the pathological sites, making it difficult to provide effective therapy at the targeted disease locations. Due to its special qualities, which include its minimally invasiveness and high spatiotemporal resolution, light‐mediated therapeutic strategies have lately become one of the most promising and effective tools for precisely controlling the activation of therapeutic reagents and imaging probes in vitro and in vivo [9].
Thus, in addition to discovering new, effective, targeted antitumour drugs, it is crucial to come up with creative ways to provide cancer chemotherapy with a more focused, targeted impact. An alternative that has long been of interest to researchers is the application of different internal triggers (such as pH, redox environment and enzymatic processes) or external triggers (such as ultrasound, magnetic field and light irradiation) for the on‐site induction of stimuli‐sensitive drug compounds or drug delivery systems [10]. Out of all the possible physical stimuli, light stands out for a number of reasons, including its exact control over the wavelength and amount of radiation, as well as its interactions with biological systems [11].
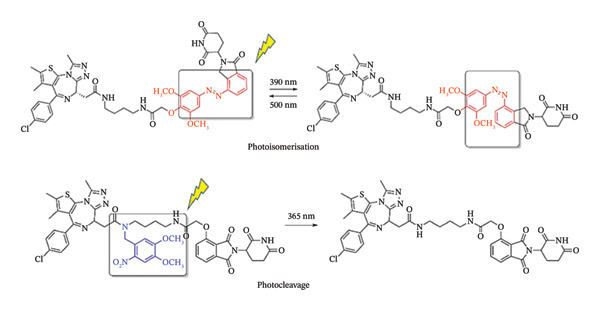
In this regard, the area may also profit from the instruments already existing for photodynamic treatment (PDT). Light activation can target a range of malignancies since it is not influenced by the characteristics of the environment within the tumour. Following the approval of the first PDT regimens in the 1990s, light‐activation‐based techniques were put into clinical use [12]. Two light‐activation‐based strategies also surfaced. As outlined by Scheme 1, photocaged vs. photoswitchable pharmacological agents are described with the respective photoactivation mechanisms. These mechanisms utilize photoswitches and photoremovable protective groups (PPGs), or “photocages” [13], an area is also referred to as photopharmacology.
Photocaged vs. photoswitchable pharmacological agents showing the respective photoactivated transformations. Adapted from [13]. Cancers, published by MDPI, 2021.
Molecules that are photocaged are inactive in the absence of light and become physiologically active only upon exposure to light [14]. The caged molecule can be released with spatiotemporal control thanks to this light activation [15]. Another way that photocaging differs from PDT is that it is not dependent on oxygen, whereas photosensitizers (PSs) often need a high oxygen concentration to function [16, 17]. The cage and the caged molecule are the two halves of a photocaged complex. The drug is released from the caged molecule when it absorbs a photon of light at the right wavelength, making the caged molecule physiologically active. This approach is referred to as photochemotherapy (PCT) when used in cancer therapies [18, 19].
Both photocaging and photoswitchable strategies are feasible for small compounds, where photoactivatable parent drugs are made by structural alterations, or for more sophisticated drug delivery systems, photoactivatable units are strategically attached into the framework [20, 21].
2. Comparative Overview of PSs in Phototherapy
There are primarily three forms of phototherapy: photodynamic therapy (PDT), photothermal therapy (PTT) and photoactivated chemotherapy (PACT). The three main elements of PDT and PACT are a PS, exposure to a certain wavelength of light and the presence of molecular oxygen (or another oxidizable substrate). In each instance, the PS absorbs a photon, undergoes photochemical processes that produce reactive oxygen species (ROS) and is transformed into an excited state (first singlet, then triplet via intersystem crossover). Target cells (tumour cells or bacteria) suffer oxidative damage as a result, which causes cell death or inactivation.
PDT is now used to treat select malignancies that are easily accessible with lamps or optic fibres, such as some forms of head and neck, cutaneous, bladder, lung, oesophageal cancers and others. It offers significant benefits, including temporal and spatial manipulation. It is additionally being intensively investigated for use in other contexts, including for the cure of bacterial and fungal illnesses that are immune to other drugs [22].
Two primary PDT mechanisms of action (Type I, which includes the generation of ROS, and Type II, in which ^1^O_2_ directly triggers the damage) depend on the PS’s ability to change oxygen from its triplet ground state to its singlet excited state. PDT typically entails the injection of a PS, which produces cytotoxic species when exposed to oxygen and is triggered by visible light. These mechanisms are shown in Figure 1 [23]. Utilizing e/H^+^ transfer mostly from PS ^∗^(T_1_) states, a Type‐I PS produces oxygen‐free radicals such as the hydroxyl radical (OH^−^) and superoxide radical (O_2_ ^−^).
Jablonski’s diagram for the principle of Type I, Type II, and Type III PDT. S0, the singlet ground state; S1, the first excited singlet state; S2, the second excited singlet state; Sn, the nth excited singlet state; IC, internal conversion; ISC, intersystem crossing. Reprinted with permission from [23] Copyright (2022), Elsevier.
Conversely, a Type‐II PS produces singlet oxygen (^1^O_2_) and PS (S_0_) by triplet‐triplet annihilation, which is fuelled by electron spin exchange between PS ^∗^(T_1_) and triplet oxygen (^3^O_2_). In other words, oxygen is necessary for Type‐I and Type‐II processes to produce ROS as intermediates. Oxygen is required for photosensitization to occur but solid tumours thrive in a hypoxic environment. This has a big impact on how well photosensitive medications kill tumour cells. As a result, much effort has been put into preventing PS reliance on oxygen‐dependent PDT [24].
Tumours, specifically in the inner core region, are frequently hypoxic. As mentioned above, it would be quite intriguing to create a light‐mediated approach that is independent of oxygen. In this way, light can have other effects besides singlet oxygen production. It is known that chemical bonds may either cleave or reorganize when exposed to light. In fact, a compound’s functionality may be changed by enclosing some of its functional groups in a photoremovable group. Then, exposure to light can regulate the original compound’s release.
A number of medications and drug candidates have been effectively released when exposed to light using photocages. These include inhibitors of carbonic anhydrase II, aspirin, ibuprofen, ketoprofen and a few anticancer drugs including floxuridine, paclitaxel, tegafur, doxorubicin and a photoreactive DNA intercalator [25]. A cleavable singlet oxygen bridge was previously used to attach the anticancer drug candidate combretastatin A‐4 to a porphyrin PS in a recent combination of photocaging and singlet oxygen production [26].
On the other hand, photothermal treatment uses metal complexes to convert light into thermal energy through nonradiative relaxation pathways. This process is referred to as the photothermal effect. Transition metal complexes represent a viable alternative for anticancer phototherapy, since they exhibit high toxicity towards cancer cells through photoreduction and photosubstitution processes. Generally, a sequence of photochemical reactions or photothermal effects are started to kill tumour cells when low toxicity metal complexes precisely concentrate in tumour areas through blood circulation and are triggered by light of specified wavelengths. For in vivo applications, photoresponsive metal complexes continue to present challenges. This is because of their poor stability in physiological settings and adverse metabolic/biodistribution. Additionally, metal complexes are quickly removed from the circulation and cannot effectively accumulate at tumour sites.
Furthermore, metal complexes may have hazardous adverse effects and are often not biocompatible. Thus, the development of metal complex‐containing materials that tackle these issues for the treatment of tumours in vivo is extremely important. In this regard, metallopolymers that are produced by integrating or incorporating metal complexes into polymeric scaffolds show promise for use in living organisms [27, 28].
3. Photoactivation UV Responsive Photocages
Light of various wavelengths is a perfect stimulus with noninvasive qualities and spatiotemporal accuracy. Generally, a photocleavable functional group that inhibits the action of therapeutic molecules is activated by a light beam, particularly in the short wavelength UV range. This allows for carefully regulated drug release in the target microenvironment.
Numerous other photocaged compounds have been developed for spatiotemporal regulation of physiological events, real‐time cell transport surveillance and regulated discharge of active compounds in vitro and in vivo. Examples of these molecules are o‐nitrobenzyl (ONB), pyrenylmethyl ester and coumarinyl ester [9, 29] which are shown in Scheme 2 that outlines the UV light‐mediated uncaging strategies for these compounds.
UV light‐mediated uncaging strategies: (a) o‐nitrobenzyl, (b) pyrenylmethyl ester, (c) coumarinyl ester, and UV light‐induced release of D‐luciferin from the ONB photocaged D‐luciferin derivatives. Reprinted with permission from [30]. Copyright 2015. American Chemical Society.
Photocaged nitrobenzene ligands, for example, have been utilized to study the firefly luciferase (fLuc) bioluminescence reporter in real time, both in vitro and in vivo. To do this, a series of stable and effective photoactivable D‐luciferin probe molecules were developed by several researchers. As illustrated in Scheme 2, the inactivation of D‐luciferin’s 6‐hydroxy unit by various nitrobenzene ligands was responsible for initially preventing D‐luciferin’s bioactivity [30, 31].
These photocaged devices, which exhibit powerful bioluminescence imaging in living mice, were quickly released to restore the activity of D‐luciferin under UV light irradiation at 365 nm [30].
Using low‐energy radiation, McCoy et al. created a molecular approach for light‐triggered drug administration of several kinds of drugs [32]. The amount of applied light accurately controls the drug dosage. The molecular unit functions as a dosage device at the molecular level when combined with the proper polymer scaffold [32]. Using a light‐controlled drug liberation reaction, the high degree of control that can be applied to light imposed to a molecule in terms of wavelength, duration, intensity and location can be utilized to give control over the amount of drug released (the dose), the exact moment of the release event and where it occurs [32].
Significantly, this control may function at the single molecule level, enabling conjugate‐incorporated media to function as molecularly controlled drug dosage devices [33]. Derivatives of 3,5‐dimethoxybenzoin (3,5‐DMB) have been employed in the past as protective groups in chemical synthesis, which may be detached by applying light [34]. This method of protecting molecules having carboxylic acid or secondary amine functional groups involves reacting them with 3,5‐DMB to produce esters or carbamates, respectively.
Wei et al. synthesized a photolabile derivative (1) of the anticancer medication 5‐fluorodeoxyuridine (2) as a model prodrug [35]. Using long‐wavelength UV light to photolyse Compound 1, Compound 2 was quickly liberated into solution (Scheme 3). Compound 1 by itself is not toxic to cells, but when combined with UV light (λ = 350 nm), it significantly inhibited the proliferation of cells. Long‐wavelength UV light photoactivated the nontoxic, photolabile Prodrug 1 into the toxic Drug 2, resulting in a strong suppression of cell development.
Photolysis of 1 with long‐wavelength UV light rapidly released 2 in solution. Adapted with permission from [35]. Copyright (2020), Elsevier.
4. Infra‐Red Responsive Photocages
The traditional approaches of light‐mediated theranostics, while initially successful, are mostly focused on short‐wavelength light (UV or visible light), which typically has a number of undesirable side effects. These involve the possibility for phototoxicity to healthy tissues, the restricted depth of tissue penetration and inevitable light absorption and dispersion [36].
Due to their exceptional optical qualities, the 4,4‐difluoro‐4‐bora‐3a,4a‐diaza‐s‐indacene (BODIPY) and its analogues are widespread fluorescent dyes that have become utilized extensively in bioimaging, disease diagnostics and PDT [37, 38]. They exhibit long nanosecond‐scale fluorescence lifespan, strong molar absorption coefficients, intense fluorescent emissions with comparatively narrow emission bandwidths and photostability, among other qualities. With these characteristics, Goswami et al. have reported that meso‐BODIPYs might serve as photocages as well [39]. They developed a computationally searching technique to show that, under green light irradiation (515–553 nm, 500 W), the meso‐position (C8) on BODIPY (II‐1, II‐2, Figure 2(a)) was prone to create carbocation with a low‐lying excited state to liberate acetic acids.
FIGURE 2BODIPY‐based photocaging structure. (a) Basic structure of meso‐methyl BODIPY‐based photocages. (b) BODIPY‐based photocages with diverse subcellular sites. (c) BODIPY‐based photocages with enhanced water solubility. Reprinted with permission from [40] Copyright (2023), Elsevier.(a)(b)(c)
Using this platform, they coupled several organelle‐targeting groups to the a‐methyl position in order to accomplish subcellular photorelease (II‐3, Figure 2(b)). Live‐cell fluorescence was seen in comparable organelles with the release of p‐nitroaniline (PNA) under the excitation light (545 nm, 42mWcm}2). These findings led to the development of a novel medication delivery method based on the spatiotemporal regulation of photouncaging [41].
Further optimization of the structures of BODIPY has been attempted by the addition of hydrophilic sulfonic acid groups without affecting their spectroscopic or photoreaction characteristics in order to increase the water solubility of such BODIPY‐based photocages [41]. In this regard, sulfonates with long linkers have been connected at different sites while electron‐donating groups (EDGs) were added to the 2,6‐positions of the BODIPY structure (II‐4, Figure 2(c)). Increased solubility and light‐dependent release of dopamine uncaging in neurons were the outcomes of this chemical designs. Thus, the cellular localization of water‐soluble BODIPY‐based photocages was completely regulated, while keeping the photoreaction properties unaffected.
5. Metal Complexes in Photocages
It has also been suggested that the use of light to treat cancer, which has significantly contributed to the clinical development of PDT, represents an intriguing advancement in the study of metal‐based anticancer drugs that do not require oxygen to function, like PSs. There are numerous publications on the utilization of photocaged organic molecules, but only few instances of the specific release of intact (organo)metallic compounds triggered by light. It is true that some complexes have light‐induced reactions in which the metal centre participates. This characteristic was utilized to create metal‐based photocages in addition to increasing their cytotoxicity. Cytotoxicity may also be induced by the release of metal ions or a labile metal complex, as demonstrated by employing a photolabile o‐nitrophenyl moiety [42].
In the initial stages of their production, photocages were mostly made of organic substances. In 1978, an ONB group was used in a pioneering instance of photocaging, which released ATP in response to UV light [43, 44]. Metal coordination complexes have been used as photocages more recently. To achieve uncaging, metal complexes go through a convoluted series of electronic transitions [45, 46]. The molecule may excite an electron from the HOMO (^1^GS) to the LUMO upon photon excitation [47]. The metal‐to‐ligand charge transfer (^1^MLCT) state is the name given to this singlet excited state.
Subsequently, the excited electron can reach a triplet excited state (^3^MLCT) by undergoing an intersystem crossover and spin inversion. The electron’s destiny inside the ^3^MLCT is dependent upon the energy levels of the accessible decay routes. The metal complexes may undergo ligand cleavage and shift into the triplet metal cantered state (^3^MC) if the ^3^MC state is thermally accessible. The ^3^MC state will then relax back to the ground state via phosphorescence or a nonradiative decay process if its energy is too high in comparison to the ^3^MLCT state [48].
6. Ruthenium (II) Metal‐Based Photocages
Many low‐spin d^6^ transition metal compounds, such as Mn(I), Re(I), Fe(II), Ru(II), Os(II), Rh(III), Ir(III) and Pt(IV), are photoactive [49]. Certain complexes may exhibit phosphorescence decay from a triplet excited state and have long‐lived excited states, based on the kinds of ligands. Others may suffer redox reactions resulting from photoinduced metal and/or ligand electron transfer, or they may undergo metal–ligand bond weakening in singlet or triplet excited states, hence promoting ligand release [50].
Historically, photocaging has been used on functional groups present in inactive compounds such as carboxylic acids, alcohols and amines [51, 52]. UV radiation is necessary for the majority of photoremovable organic protective groups to uncage, which negatively affects biological systems by causing irreversible photodamage. Moreover, organic groups do not shield functional groups which are unprotectable by organic molecules. These groups include nitriles and aromatic heterocycles, which are crucial “warheads” that engage in direct interactions with heme and thiolates in the active regions of protein targets [52].
For caging applications, transition metal fragments are appealing, particularly complexes that attach to functional groups that are unprotectable by organic molecules. Therefore, an alternative method for photocaging bioactive compounds is provided by metal coordination. Metal‐based photocaging makes it easy to release bioactive molecules when exposed to low‐energy light since transition metal complexes frequently show strong charge transfer absorption in visible wavelengths [48, 53], and the bond between the metal and its related ligand is nearly always less stronger compared to an organic sigma bond [54]. One may adjust the photochemistry to achieve drug release throughout a wide spectral range, from the visible to near‐infrared (NIR) frequencies, by adjusting accessory ligands.
In this regard, metal‐caged compounds are intriguing chemical substances for biological investigation and applications because they can be spatiotemporally controlled to free them selectively. Metal‐based photocaging is currently being used to create innovative light‐activated therapies in addition to other fundamental research uses [13, 55].
Photocages made of ruthenium(II) polypyridyl complexes have been synthesized and reported by several researchers. Ru(II) complexes’ octahedral shape allows for excited state ligand dissociation, a property that is not available for square planar Pt(II) such as cisplatin [56]. Ru(II) polypyridyl complexes can absorb in the visible spectrum and are generally thermally inert in aqueous solutions [57]. Some Ru(II) complexes concentrate at tumour cells because of their ability to imitate iron binding [58, 59] which is a desirable feature for drug administration. After light is absorbed, the thermal population of 3d‐d states transitions from a triplet ^3^MLCT state leads to ligand dissociation in many Ru(II)‐based polypyridyl complexes.
Metal complexes made of ruthenium are highly valued for their facile singlet oxygen generation and significant absorption in the visible spectrum [60]. They may be adjusted for certain therapeutic uses and frequently show high photostability [61]. Because of their great degree of tunability, ruthenium(II) polypyridyl complexes may be designed to absorb light in the visible (400–600 nm) or even NIR spectrum by modifying their ligands. This is the case for [Ru(bpy)_2_L]^2+^ complexes (where bpy = 2,2′‐bipyridine), whereby, when exposed to radiation, they can release bioactive ligands. The ability to absorb light is beneficial for deep tissue penetration, which is a significant drawback for many PDT medicines. As effective light‐responsive agents, Ru(II) complexes proceed through MLCT or photoinduced ligand dissociation processes.
A class of inhibitors that consist of nitrile‐containing compounds reacts with the thiolates found in the active site of cysteine proteases thereby inactivating them [62–64]. The nitrile functions acts as the “warhead” of these compounds, forming a covalent link with the target protein [65, 66]. For instance, the nitrile‐based cathepsin K inhibitor odanacatib, which has progressed to Phase III clinical trial for the treatment of osteoporosis, binds to the target enzyme’s active site and decreases bone resorption [67, 68]. It has been established that cysteine proteases play crucial roles in the emergence of cancer [69, 70]. Cysteine protease suppression, therefore, has anticancer effects via preventing metastatic growth and invasion, as well as by preventing enzyme‐substrate interactions [71]. Cysteine protease inactivation only happens under light irradiation since nitriles may be securely coupled to Ru(II).
In this regard, Ru(II)‐based protecting groups have been employed to cage known nitrile‐based cysteine protease inhibitors in order to modulate cysteine protease inhibitions both spatially and temporally with the caged antagonists exhibiting the capacity to be liberated upon exposure to light.
Each of the antagonists has an electrophilic nitrile group that can interact with cysteine proteases’ target sites’ thiolate to block the enzyme. The photochemical characteristics of their Ru(II)‐caged complexes are typically similar. Figure 3 indicates the process of photoinduced ligand exchange in Ru(bpy)2‐caged nitrile‐based cysteine protease inhibitors. In this mechanism, they remain stable in aqueous media under dark conditions. However, when exposed to visible light, they successfully discharge the caged drugs, usually releasing both of the two equivalents (Figure 3) [52]. All caged compounds display a substantial increase of cysteine protease inhibition when exposed to light, with each light‐activated complex exhibiting a stronger effect on cysteine protease inhibition compared to inhibitor on its own [52].
Schematic representation of photoinduced ligand exchange in Ru(bpy)2‐caged nitrile‐based cysteine protease inhibitors. Reprinted with permission from [52] Chemical communications (2028), Royal Society of Chemistry.
Treatment of [Ru(bpy)2 [1]2]^2+^ with light irradiation results in the B32‐fold improvement of the inhibition of cysteine protease papain in comparison to dark experiments [52]. Papain‐based control studies using [Ru(bpy)2(MeCN)2]^2+^ rule out the potential for enzyme inhibition brought on by other photolysis products, suggesting that the Inhibitor 1 produced in response to light irradiation is the source of enzyme inhibition [52].
The luminescent 4d^6^ Ru(II) complex TLD1433 (Ruvidar®, also referred to as Rutherrin) (Figure 4), which has undergone clinical trials for the management of bladder cancer, is activated by green light causing it to produce ^1^O_2_. It is water soluble and characterized by high ROS generation. It is known to preferentially accumulate in bladder tumours targeting transferrin receptors (TfR).
Chemical structure of TLD‐1433 and its NIR‐activated mechanism of action. Reprinted with permission from [72] Copyright (2022), Elsevier.
In contrast to TLD1433’s PDT effects, 5d^6^ Pt(IV) complexes frequently break down directly to produce cytotoxic species even in the absence of oxygen [73]. This property makes them potentially useful in PACT, which includes phototherapy for hypoxic malignancies.
Modifications to the structure of tris N^N Ru complexes are widely recognized approaches where, for example, two complexes with the structure [Ru(phen)2(7R,8R‐dppz‐)]^2+^ (R = OH or ‐OMe) were reported by the Gasser group [74]. Intense phosphorescence and high ^1^O_2_ quantum yields were demonstrated by both complexes, but their cellular accumulation and toxicity were different. It is possible to fine‐tune PDT characteristics through modifications of the intercalating groups, as demonstrated by the various toxic effects reported for each complex before and after irradiation (at 420 or 800 nm for monolayer complexes or spheroids, respectively) [75]. Different classes of these complexes frequently exhibit uneven trends, which can lead to misunderstandings. For instance, it was once believed that the presence of bulky groups near the metal centre increased photodissociation and consequent toxicity. Nevertheless, studies conducted under acidic and basic conditions revealed that deprotonation decreased dissociation and toxicity in bis‐heteroleptic N^N complexes with ionizable groups positioned to put a strain on the coordination sphere [76]. Comparing deprotonated complexes to their protonated counterparts, higher ROS quantum yields were observed, indicating that phototoxicity in the complex is caused by ROS rather than photodissociation [76].
7. Ruthenium PACT Compounds
Microtubules are crucial for the separation of double chromosomes prior to cell division during mitosis. Because of this characteristic, they are a good target for new chemotherapeutic drugs, which selectively destroy rapidly dividing cells. Paclitaxel is one of the most effective microtubule targeting drugs used in clinical settings. It is utilized for the treatment of several types of nonsmall cell lung cancer, ovarian cancer and breast cancer. Paclitaxel causes apoptosis and cell death by interfering with the dynamics of microtubules. However, adverse effects from paclitaxel‐based treatment are common and involve neurotoxicity, neutropenia and irregular heart rhythms, among other things. Furthermore, the clinical effectiveness of paclitaxel chemotherapy is limited due to cancer cell tolerance. Therefore, an important field of cancer research involves developing alternative microtubule‐targeting drugs or techniques.
When compared to their ability to function as PSs, the field of Ru(II)‐based photocages is far less established. There have been important discoveries in the recent past that show the wide range of beneficial applications for this technology [77–80]. The first instance of uncaging a neurotransmitter from a Ru(II) complex was accomplished in 2013 by the Etchenique group (Figure 5(a)) [81, 82]. A Ru(II) photocage complex demonstrating the first instance of light‐activated protease inhibition was reported in 2011 (Figure 5(b)) [81].
FIGURE 5Ru(II)‐based photocage reactions. Adapted from [81].(a)(b)(c)
The intense absorption of visible light into singlet ^1^MLCT states, which undergoes intersystem crossing with almost 100% yield to fill corresponding triplet MLCT (^3^MLCT) states, is a key characteristic of Ru(II) complexes [83, 84]. When a ligand is coordinated with an extended π‐system that has a ligand‐cantered π‐π ∗ state that is below the ^3^MLCT state, long excited‐state lifetimes (20–50 μs) are generated [85, 86]. The production of singlet oxygen (^1^O_2_) is the mechanism of action for the majority of Type II PSs used in clinical PDT.
Significant progress has been achieved subsequent to these foundational works. A Ru(II) photocaged complex irradiated with green light has been shown to release a drug that targets microtubules, in contrast to the previously mentioned examples which were uncaged by the use of blue light of higher energy [87, 88].
Moreover, this derivative of a photocaged system was the first Ru(II) PCT agent to be triggered in an in vivo mouse model—a unique outcome that was crucial to the advancement of this discipline (Figure 5(c)) [88].
Moreover, complexes with sterically hindering ligands that deviate from the ideal octahedral structure in the arrangement around the Ru(II) centre produce lower energy metal‐centred states of antibonding character that can be populated from ^3^MLCT states [89, 90]. This makes ligand disengagement easier and is a notion that is utilized in PCT. Ru(II)‐based complexes with dual functionality that are accessible with low‐energy illumination have been reported by several researchers. These complexes can be used as drug models since they can produce ^1^O_2_ through photosensitized photosynthesis and cause aromatic heterocycles to photodissociate [81]. A crucial component of this dual action PDT/PCT drug was the π‐expansive ligand 3,6‐dimethylbenzo[i]dipyrido[3,2‐a:2′,3′‐c] phenazine (Me_2_dppn), which is composed of a phenanthroline core linked with a diaminonaphthalene moiety.
Significantly, it was demonstrated that effective, light‐activated cell killing in in vitro models of triple‐negative breast cancer was achieved through the combination of PCT and PDT with a Me_2_dppn‐containing Ru(II) complex [81]. In these 2D and 3D culture models, related complexes that underwent PCT or PDT alone exhibited little to no cytotoxicity, indicating that combined action PCT/PDT generates and enhances the effectiveness of Ru(II) compounds against cancer cells.
Compared to conventional chemotherapy, PDT has been demonstrated to cause less systemic damage in cancer patients because light is administered precisely to the tumour environment. Nonetheless, PDT and PACT vary fundamentally from one another. Through the energy or electron transfer of the excited prodrug molecule to the O_2_ present in the irradiated tumour tissues, a light‐irradiated PS in PDT produces cytotoxicity [91, 92]. PDT is therefore very effective in cancers with adequate oxygenation, but it frequently fails in hypoxic tumours because low O_2_ concentrations prevent oxidative stress from being produced by photolysis [93–96].
Quite the reverse, PACT relies on an O_2_‐independent bond cleavage photoreaction for its light stimulation process, which means that PACT should theoretically enable the development of anticancer phototherapies that do not rely on intratumoural oxygen levels and can thus be used for treating hypoxic tumours. An important goal in oncology is to come up with novel therapies that are safe in the dark, that upon irradiation with light concentrate on a well‐established cancer target and that remain effective under hypoxic environments. This is because oxygen‐deficient tumours are highly challenging to treat by PDT, radiation therapy, chemotherapy or immunotherapy [97, 98]. Studies have examined and reported the use of 22 as a novel PACT complex that releases rigidin Compound 1 that targets microtubules when exposed to green light [99].
The studies provided quantitative phototoxicity experiments in vitro, showing effective light activation at low O_2_ concentrations under normoxia (21% O_2_) and hypoxia (1% O_2_) [88]. Furthermore, it is demonstrated for the first time in 3D tumour spheroids and human lung cancer (A549) xenografts in nude mice the light activation of a ruthenium‐based PACT complex. Thioether‐containing microtubule Inhibitor 1 was reacted with two equivalents of Ru(tpy) (bpy) (OH_2_)2 (32 (Scheme 4) to create the complex Ru(tpy) (bpy) [1]2 (22) [88].
Synthesis of [2] (PF6)2. ∗The A6 proton of the 2,2′‐bipyridine (bpy) ligand [88].
When 22 was dissolved in acetonitrile and exposed to green (λ irr = 530 nm) light in an inert dinitrogen environment, the wavelength of absorption at 452 nm increased significantly, and the maximum of the singlet metal‐to‐ligand charged transfer (1MLCT) band was slightly shifted from 452 to 454 nm [88]. When the solution was subjected to light irradiation, mass spectrometry (MS) revealed peaks at m/z = 266.4 corresponding to [Ru(tpy)‐(bpy) (CH_3_CN)]^2+^ (calcd m/z = 266.1). In contrast, a strong signal was seen at m/z = 447.0 corresponding to [2]^2+^ (calcd m/z = 447.1). This indicated that Ligand 1 was photosubstituted by a solvent molecule when exposed to green light [88].
8. A Comparative Analysis of the Biocompatibility, Cytotoxicity and Photoresponsiveness of Ruthenium (II) and Platinum‐Based Complexes
The photoresponsiveness, cytotoxicity and biocompatibility of these photocaged ruthenium (II) and platinum complexes can be summarized in Table 1. This outlines the factors in terms of modes of action, wavelengths of interest, biocompatibility and delivery profiles among others.
Photorelease therapeutics such as NAMPT inhibitors, microtubule inhibitors, metabolic inhibitors and enzyme inhibitors have excellent preclinical promise in a variety of Ru(II) polypyridyl complexes (cells and some animal models). These are only preclinical; although the area is active, human trials have not yet been implemented. Clinical challenges with translation have already been discussed in other reviews [118]. These include pharmacology issues, safety concerns, formulation challenges, tissue penetration of activating light and administration.
9. Structure–Activity Relationships of Ruthenium(II) and Platinum Photocages
As would be expected, the cytotoxic profile, tumour‐targeting potential and photoactivation effectiveness of ruthenium(II) and platinum‐based photocaged complexes are all significantly influenced by the ligand structure. Modifications to the ligand type, coordination geometry or metal centre can impact the complexes’ susceptibility to photoactivation, therapeutic qualities, including subcellular targeting, light absorption, activation mechanism, biological interactions, solubility and stability. These factors can be utilised to carry out rational drug design for these complexes by understanding the structural, electronic and steric factors that affect the activity of these complexes.
A longer wavelength (visible/NIR light) can be used for activation with electron‐donating ligands since electron‐donating ligands reduce the MLCT energy [119]. Triplet‐state durations and ROS production are increased by π‐accepting ligands, such as phenanthroline, which improve intersystem crossing [120]. Through destabilization of the ground state, bulky ligands can hasten ligand dissociation. Factors like trans‐influence acts to weaken bonds which are directly opposite to a trans directing ligand, which can hasten ligand dissociation. Photoactivation can be accelerated by substituting monodentate ligands with bentate ligands, which frequently result in more stable complexes that are slower to photodissociate [79]. For instance, substituting 4,4′‐dimethyl‐2,2′‐bipyridine for one bpy in [Ru(bpy)_3]^2+^ changes the absorption maxima and improves photolability [121].
In platinum (IV) complexes, for example, the activation of the axial ligands requires photolabile groups such as nitrophenyls or azides. By enhancing orbital overlap, flexible ligands may enable more effective photoreduction. Steric effects can be enhanced by adding aromatic rings or halogens, which can improve spin–orbit coupling and increase photoreactivity [122]. The structure activity relationships are briefly summarized in Table 2, which addresses the effects of various functionalities like steric effects, ligand geometry and electronic effects among others.
Table 3 summarizes the structural and ligand factors that influence photoactivation efficiency, cytotoxicity and tumour‐targeting ability, with the corresponding electronic effects.
As outlined in Tables 2 and 3, it can be seen that the photoactivation characteristics and therapeutic efficacy of photocaged metal complexes are greatly influenced by structural alterations. Understanding how variations in ligand types, coordination geometries and metal centres influences these characteristics have been made possible by both theoretical computations and experimental research.
For example, experimental work on platinum(II) photocaged complex [Pt(cage)] was described by Ciesiensk and colleagues. In their work, the photocaged complex had a photolabile nitrophenyl group integrated into the backbone of a tetradentate ligand that had two pyridyl and two amide nitrogen donor sites. As outlined in Figure 6, light activation induced bond cleavage, converting the neutral [Pt(cage)] 2 into a charged and exchange labile Pt(II) complex 3. When activated with UV light, the ligand backbone was broken, producing a Pt(II) complex that more easily exchanged its ligands with a peptide containing methionine groups. Prior to this, the intact complex was insensitive to ligand‐exchange processes [126].
Light activation inducing bond cleavage, converting neutral [Pt(cage)] 2 into a charged and exchange labile Pt(II) complex 3. Adapted from [126].
In the dark, the complex had no effect on MCF‐7 breast cancer cells, however short‐term exposure to UV light caused the MCF‐7 cells to die at a rate that was comparable to cisplatin [126]. This is a new approach that might possibly deliver metal‐based medications in a site‐specific and time‐specific way by utilizing light to change the coordination chemistry surrounding the metal core.
9.1. Photoresponsive MPNs of Platinum
Pt‐based anticancer medicines, the first class of therapeutic anticancer metal complexes, are utilized to treat more than 40% of cancer victims because of their potent interactions with DNA nucleobases, which ultimately cause cell death. Nevertheless, their hazardous side effects and modest tumour accumulation continue to impede their clinical advancement. As a result, therapy dependent on Pt anticancer drugs is unable to produce steady therapeutic benefits.
Typically, Pt(II) complexes need UV light (λ < 400 nm) to activate, which has poor tissue penetration and can harm nearby tissues [127]. Although ligand loss or the oxidative production of ROS is typically involved in their photoactivation, these mechanisms are frequently less effective than in Ru(II) systems. Pt(IV) prodrugs release active Pt(II) species when activated by light, but they need to be structurally optimized for visible and NIR responsiveness.
Due to their high cytotoxicity when activated and ability to lead to the formation of DNA cross‐links, platinum complexes, particularly those based on platinum (IV), have been evaluated as a substrate for PDT [128]. Because of these qualities, they are highly effective killers of cancer cells. Nevertheless, the overwhelming toxicity of platinum‐based PSs presents highly pertinent issues associated with the biocompatibility and safety. Moreover, they are typically less photophysically favourable compared to their analogues and iridium and ruthenium complexes, especially for light harvesting and ROS generation efficiency.
Pt‐containing MPNs, on the other hand, may considerably increase therapy efficiency by enhancing the tumour accumulation impact and minimizing hazardous adverse effects to normal tissue.
Many Pt‐containing MPNs have been created for in vivo and in vitro anticancer phototherapies [8]. Since Pt(IV) azide‐based octahedral MPN compounds have lower spin d^6^ compared to the d^8^ configuration of their Pt(II) counterparts, they are less susceptible to replacement [129–131]. Additionally, Pt(IV) complexes can be rendered photoactive by selecting appropriate ligands, such as the photolabile azido group [132, 133]. Indeed, azide complexes of transition metals are often light‐sensitive. Trans‐[Pt(N_3_)2(CN)4]^2−^, which possesses a MLCT electronic absorption band (Pt ⟶ N_3_) at 302 nm, is the first photoactivated Pt(IV) complex containing azide ligands [134].
MPNs have been used to administer photosensitive Pt(IV)‐azide prodrugs in conjunction with other medicinal substances, in addition to their use in single‐drug photochemotherapy. There are at present two main ways that combinations are formed: either by entrapment or by linking covalent bonds to the Pt(IV)‐azide prodrugs. In order to create a multifunctional drug conjugate that can be further grafted to the side chains of copolymers, some medicines can first work in concert with Pt. Secondly, the core of MPNs can also enclose hydrophobic medicines. Chemotherapy with multidrug combinations can enhance the effectiveness of anticancer therapies by stimulating several signalling pathways in cancer cells. A photoresponsive Pt‐containing polymer (P‐Z‐DMC‐CIS(N_3_)) for multidrug chemotherapy was described by Zhou and colleagues (Figure 7(a)) [135].
(a) Preparation of P‐Z‐DMC‐CIS(N3) and P‐Z‐DMC‐DXA(N3) micelle. (b) The working mechanisms of P‐Z‐DMC‐CIS(N3) micelle in cisplatin‐resistant cancer cells. Reprinted with permission from [135]. Journal of Materials Chemistry B. Royal Society of Chemistry (2015).
The derived P‐Z‐DMC‐CIS(N_3_) had the capacity to discharge active Pt(II) and DMC inside acidic endosomes/lysosomes and was sensitive to light treatment (365 nm, 10 mW·cm^−2^). Essentially, repair of nucleotide excision and defence triggered by DNA damage requires the widely expressed protein phosphatase 2A (PP2A) (Figure 7(b)) [135]. P‐Z‐DMC‐CIS (N_3_) showed synergistic therapeutic benefits for cisplatin‐resistant cells (A549R) with a half maximum inhibitory concentration (IC50) of 8.4 μM, thanks to improved cellular absorption and photocontrolled discharge of the integrated medicines [135].
In a different instance, oxaliplatin (IV) complex and DMC were combined to create a drug conjugate, which was then grafted to the same polymer carrier to create P‐Z‐DMC‐DXA(N_3_) [136]. When used against HeLa cancer cells, the polymer nanoparticles had a remarkable synergistic effect in inhibiting cancer cell development. In addition, the P‐Z‐DMC‐DXA(N_3_) showed a positive anticancer impact, as the tumour volume progressively shrank from 100 mm^3^ to 40 mm^3^. More significantly, following P‐Z‐DMC‐DXA(N_3_) therapy and UV light irradiation (365 nm, 10 mW·cm^−2^), there was a 100% survival rate [136].
He and colleagues created a different kind of MPNs for multidrug combination treatment. Dextran‐Pt(N_3_) conjugate polymer nanoparticles (Cur@DPNs) were loaded with the photosensitive anticancer medication curcumin (Cur) (Figure 8(a)). The release of active Pt(II) and the generation of ROS were aided by the photoactivation of Cur@DPNs caused by 365 nm UVA light, which significantly reduced the viability of cancer cells (Figure 8(b)) [137]. Their results as demonstrated in Figure 8(b) demonstrate the intracellular process after endocytosis of Cur@DPNs for combinational photoactivated therapy. The overall observation is that combinational photoactivated therapy was superior to the previously reported methods of single PACT. Identifying the manner and the time frame that polyprodrug nanoparticles are photoactivated and dissociated to release active drugs in cancer cells is the primary challenge to nanoparticle‐mediated drug delivery [122].
FIGURE 8(a) Synthesis light stimulation and dual–drug sensitivity of Cur@DPNs and (b) illustration of the intracellular process after endocytosis of Cur@DPNs for combinational photoactivated therapy. Reprinted with permission from [137] Copyright, 2016. American Chemical Society.(a)(b)
The O_2_ consumption during PDT aggravates hypoxia within a solid tumour which greatly reduces the production of ROS and further undermines the PDT efficacy. Despite one‐electron transfers from two hydroxyl groups to the Pt(IV) centre, the Pt (IV)‐azide complexes with cis‐diamine ligands could be light‐triggered to generate Pt(II) species and hydroxyl radicals that readily dimerize to hydrogen peroxide (H_2_O_2_) [138].
Under UV radiation, H_2_O_2_ then breaks down into ordinary oxygen and water. Consequently, the administration of PSs in conjunction with oxygen‐self‐generating Pt(IV) prodrug is beneficial for mitigating tumour hypoxia and promoting synergistic anticancer action. This was demonstrated by Xu and colleagues, by coupling PEG‐modified PS chlorin e6 (Ce6) to Pt(IV)‐azide complexes with cis‐diamine ligands. In order to create UCNPs‐embedded nanostructures (UCPP) with a mean size of 60 nm, the polymer was subsequently coassembled into upconversion nanoparticles (UCNPs). Figure 9 showa a representation of the light‐driven separation of UCPP in tumour microenvironment. The figure illustrates the formation of UCPP and its light‐driven dissociation including O_2_ production, simultaneous activation of PDT, and active Pt(II) liberated for the synergistic photochemotherapy [139].
Representation of the light‐driven separation of UCPP in tumour microenvironment illustrating the formation of UCPP and its light‐driven dissociation including O2 production, simultaneous activation of PDT and active Pt(II) liberated for the synergistic photochemotherapy. Adapted from [139].
Other compounds explored as NIR‐responsive candidates are Pt(IV) complexes. These compounds may break down and produce oxygen when 980 nm NIR light has been transformed by the UCNPs into shorter wavelength emissions of between 660 nm and 365 nm. The ROS self‐generating ability of UCPP remained unaltered even in a hypoxic environment under 980 nm irradiation, which reversed PDT‐induced hypoxia and increased PDT efficacy, owing to the high O_2_ level [139]. In both normoxic and hypoxic environments, L929 cells were considerably more susceptible to UCPP light exposure than the single‐function reference group. Furthermore, the fluorescence signal at the tumour location grew over the course of 24 h, and the drug’s in vivo bio distribution at several organs verified the precise build‐up of UCPP in the tumour cells [139].
Following irradiation with 980 nm laser treatment, UCPP demonstrated considerable tumour suppression efficacy with total tumour eradication in the HeLa, HCT116, and MDA‐MB‐231 tumour‐bearing animals [139]. The majority of additional haematoxylin and eosin (H&E) stain fragmentation regions and the fewest positive tumour cells detected by proliferating cell nuclear antigen (PCNA) expression were also noted. These findings showed that under NIR irradiation, UCPP may self‐generate Pt(II) and O_2_. This new photosensitive metallopolymer nanoplatform presents a promising method for mitigating hypoxia in tumours and enhancing the therapeutic impact of phototherapy.
10. Porphyrin‐Based MPNs of Platinum
In solid tumours, the aggressive growth of tumour cells and inadequate oxygen delivery lead to a severely hypoxic microenvironment. In this sense, the degree of hypoxia may serve as a gauge for the magnitude of the tumour. Consequently, the development of dual‐functional MPNs for combined PDT therapy and hypoxia detection is an attractive area. In this regard, Pt(II) porphyrins have garnered a lot of interest, where they are recognized for their significant stokes shift, extended triplet excited state lifetime and photostability as a typical phosphorescent transition‐metal complex (PTMC).
Zhou and coworkers conducted a ground‐breaking study wherein they utilized Suzuki coupling polymerization to include Pt(II) porphyrin into a hyperbranched linked polyelectrolyte that was initially insensitive to oxygen (Figure 10(a)) [140]. The polyelectrolytes could build up in tumour cells through the EPR effect as they self‐assembled into polymer dots (Pdots) with a mean size of around 10 nm. Pdots’ ^1^O_2_ quantum yield reached 0.80 (λ ex = 532 nm) as a result of intramolecular Forster resonance energy transfer (FRET) increasing light absorption from the conjugated backbone to the acceptor Pt(II) porphyrin [141]. In addition, the polymer’s three‐dimensional structure helped to lessen self‐aggregation and undesirable intermolecular π‐π stacking. Furthermore, ratiometric O_2_ recognition was made possible by the FRET from polyfluorene units to Pt(II) porphyrin [141].
FIGURE 10(a) Chemical structure, TEM image of polymer dots (Pdots) in PBS solution and O_2_‐sensing mechanisms during PDT of the Pt(II) porphyrin‐containing Pdots [140]. (b) Chemical structures of Por(Pt)‐bisUPy and DPA‐bisUPy. Reprinted with permission from [141]. Materials Chemistry Frontiers. The Royal Society of Chemistry (2018).(a)(b)
Analysis of apoptosis and necrosis in HepG2 cells revealed that polymer dots had minimal dark cytotoxicity and that the ROS they produced was what caused the cell mortality to accelerate as the exposure period increased [142, 143]. This shows that PDT therapies and intracellular hypoxia recognition are feasible at the same time, and it offers vital guidance for the creation of phosphorescent diagnosis‐therapy integrated systems in the future [140].
Huang et al. assembled a quadruple hydrogen‐bonded supramolecular polymer to create another bifunctional Pt(II) porphyrin MPN for ratiometric hypoxia detection and PDT. Their procedure is illustrated in Figure 10(b), which shows the chemical structure, TEM image of polymer dots (Pdots) in PBS solution and O_2_ sensing mechanisms during PDT of the Pt(II) porphyrin‐containing Pdots [141]. The oxygen‐insensitive fluorescence reference moiety carrying bis‐UPy served as the chemotherapeutic energy absorber in this approach, while the oxygen‐sensitive Pt(II) porphyrin group holding ureidopyrimidinone (bis‐UPy) served as the energy source. The MPNs have a straightforward synthesis and are biodegradable due to the noncovalent linkage between the monomers. The uniformly tiny particle size of the MPNs (96 nm) allowed them to easily disperse in aqueous solutions and enter tumour cells. The ^1^O_2_ quantum yield (λ ex = 532 nm) was found to be 0.70, indicating that MPNs have a significant anticancer therapeutic potency [141].
A summary of some of these complexes in various experimental stages is tabulated in Table 4. These outline the progress in research for these compounds with the mechanisms explored.
11. Organic Photoswitches and PPGs
PPGs were first employed in experiments for biological research in the 1970s [154]. As illustrated in Scheme 5, PPGs are designed with the intention of masking a substrate’s bioactive moieties by covalently attaching the protective group to a molecule that is essential to the action (a process known as “caging”) [155]. These PPGs are some of the most exploited photoswitches in biological applications. They undergo E/Z photoisomerization and photocyclization/ring opening.
SCHEME 5Overview of the most exploited photoswitches in biological applications. (a) E/Z photoisomerization‐based molecular photoswitches. (b) Photocyclization/ring opening‐based molecular switches. Adapted from [155] Doctoral dissertation, Universidad de la Rioja, 2021.(a)(b)
By eliminating the PPG with light irradiation – that is, by converting the absorbed energy into a photocleavage process known as “un‐caging” – the activity may be reinstated on demand and in a spatiotemporally controlled way [44]. In this instance, the photoactivation is irreversible, which is frequently viewed as a drawback of the methodology [156]. Even if a stoichiometric quantity of the PPG would also be present in the medium, there is still a chance that the released active agent(s) would have undesirable effects upon diffusion or excretion, generating further safety concerns [157, 158].
The research area of light‐responsive (small) drug molecules faces many of the same difficulties as other drug delivery systems, such as tissue permeation, transportation to and accumulation at the active site, control over activation signal in vivo, materials and instrumentation complexity, available translational models, and safety.
The frequency of the light needed for tissue penetration, and consequently, photoactivation, presents a unique challenge. Longer wavelength absorption, for example, often results in quicker thermal relaxation in the field of azobenzene photoswitches, a family of chemical compounds that, when exposed to light, easily isomerize between different geometries as shown in (Scheme 6) [159–162]. Moreover, the relative stabilities of the Z and E isomers may also change.
Structures of trans- and cis‐azobenzene. (a) Chemical structure of the diiodoacetamide azobenzene photoswitch, with electrophilic groups at two ends for covalent linkage to peptides. (b) Schematic diagram for the control of the peptide structure upon isomerization of the photoswitch. Reprinted with permission from [162]. Organic and Biomolecular Chemistry, Royal Society of Chemistry, 2018.
This makes probes that operate at higher wavelengths (red, infrared) or, preferably, in the biological window (λ = 650–1450 nm) advantageous, and a lot of work has gone into creating photoactivatable units in these longer wavelength ranges. A common strategy to move the absorption to longer wavelengths is to set up push–pull systems with EDG and electron‐withdrawing group arranged strategically.
In addition to chromophore modifications, certain formulations may facilitate the utilization of longer wavelength light. For example, employing UCNPs or different light sources as the Cherenkov radiation (i.e., a design predicated on internal co‐localization of the light source and the photoactive agent) could be one such formulation. Currently, a significant obstacle is that most papers discussing photoactivatable probes rely on in vitro tests (proof‐of‐concept research). These shows how applicable the idea is in general. Gaining more understanding of how these systems could function in vivo would be crucial for potential therapeutic uses. Restoring eyesight is one of the most researched areas for in vivo use of photoactivatable drugs to date [163].
12. Organic Versus Metal Photocages
Although organic compounds have long been used as photocages, they do have some significant disadvantages. Many organic photocaging units need non‐bioorthogonal UV light in order to generate photorelease [81]. On the other hand, metal photocages have the ability to release nitriles and aromatic heterocycles, which are known to be significant moieties in heme‐ and protein‐binding interactions. They also exhibit the capacity to go through electronic transitions required for uncaging at longer wavelengths [164]. This is made possible by the weaker bonding interactions between metal centres and their ligands (compared to the stronger organic σ‐bonds). Overall, these properties in metal photocages contributing to greater ease of photodissociation provide advantages over the traditional organic photocages.
Moreover, metal complexes provide the fascinating possibility of easily adjusting the overall photophysical characteristics of the complex by modifying the structures of the auxiliary ligands. By adding steric bulk to the bipyridyl ligand, for instance, [Ru(tpy) (bpy) (py)]^2+^ can be transformed into an effective photocage [Ru(tpy) (Me2bpy) (py)]^2+^ [164, 165]. This tactic is known to upset the coordination complex’s octahedral geometry and stabilize its 3MC state, which facilitates the complex’s transition from the 3MLCT into the dissociative 3MC state. Therefore, adding steric bulk to auxiliary ligands of a metal‐centred complex is a commonly used method to create effective and dependable Ru(II) photocages.
12.1. Synergistic Effects From Photoactivatable Inorganic Complexes With Immunotherapy and Gene Therapy
Due to tumour heterogeneity and adverse treatment resistance, single formulations seldom achieve the intended therapeutic effects in clinical trials [166]. There has been an increasing interest in synergistic combination treatments, which combine two or more therapeutic modalities. Gene therapy is a multipurpose treatment for cancer that can induce apoptosis in cancer cells, prevent carcinogenesis or metastasis, downregulate heat shock proteins, and stimulate the immune system by producing cytokines [167].
A possible method for improving the effectiveness of cancer treatment is the combination of immunotherapy, gene therapy, and photoactivatable inorganic compounds [168]. This strategy makes use of the systemic advantages of gene and immunotherapies as well as the exact control of phototherapy to provide synergistic effects on malignancies. By using multifunctional modes of action, the objective is to increase selectivity, decrease adverse effects, and maximize synergistic benefits [169]. Tumour‐associated antigens (TAAs) and danger‐associated molecular patterns (DAMPs), which excite dendritic cells and activate cytotoxic T lymphocytes, are released when phototherapy induces Immunogenic Cell Death (ICD) [170]. The body’s immunological reaction against malignancies is strengthened by this process.
Due to its superiority in therapeutic enhancement and reducing adverse effects, gene therapy is currently a useful adjunct to PTT/PDT [171]. There are two primary methods via which gene therapy and PTT work in concert. Either gene therapy and PTT work together to kill tumour cells, allowing gene therapy to kill nearby or PTT‐resistant tumour cells, or gene therapy can increase tumour cells’ susceptibility to PTT by, for example, preventing the production of heat shock proteins, which increases PTT’s effectiveness [171]. Gold nanoparticles (GNs), carbon‐based materials, black phosphorus nanosheets (BP NS) and other inorganic materials have all been used in gene/photothermal combination treatment [172]. Certain inorganic nanomaterials must be modified in order to load therapeutic nucleic acids; for example, the surface of the material may need to be modified with a cationic polymer to load the nucleic acid by electrostatic contact with the nucleic acid [173].
13. Recent Advances in Photocaged Compounds
Recent advances in this field have proved it to be a promising area in cancer therapy. Most notable research has been carried out by Guangyu Zhu and coworkers, who have come up with ground breaking findings in the applications of light‐activable compounds in disease treatment. They worked on customized precision medicines in cancer treatment with distinct mechanisms of action for overcoming drug resistance. Here, they came up with targeted anticancer prodrugs with controlled activation. Their prodrug named coumaplatin was a photocaged Pt(IV) prodrug based on oxaliplatin that achieved both nuclear accumulation and “on‐demand” activation as illustrated in Figure 11 [174]. The Pt(IV) complex on which this prodrug was based was found to be effectively photoactivated by water oxidation without the need of a reducing agent [174]. The complex was found to be stable in the dark but effectively activated upon irradiation with low‐intensity visible light.
FIGURE 11(a) Coumarin and (b) the proposed photoactivation mechanism of a coumarin analogue (Complex 3) in PBS buffer (pH 7.4). Reprinted with permission from [174].(a)(b)
When the prodrug was photoactivated, it showed a degree of photocytotoxicity which was up to two orders of magnitude greater than that of oxaliplatin, and was also found to accumulate extremely quickly in the nucleoli [174]. Surprisingly, this prodrug had a markedly improved capacity for tumour penetration and employed a unique mechanism of action to overcome drug resistance and cause immunogenic cell death, p53‐independent cell death and cell senescence in addition to T cell activation [174].
Their results not only offer a new approach to the rational drug design of nucleolus‐targeted and controllably activated Pt(IV) anticancer prodrugs but also show that delivering traditional platinum medications to the nucleus is a workable method of altering their canonical mechanism of action and reduce resistance.
14. Challenges and Shortcomings With Photoactivation
Despite the prospects held by photoactivation approach in cancer therapy, the development and application of photocaged medications continue to present a number of challenges. Most of the time, challenges in solubility require the usage of other molecules, including nanocarriers, to make them useful in therapy [175, 176]. Dark toxicity, or toxicity in the absence of light, is something that the perfect PS should not have. The ideal illumination should cause little off‐target retention and accumulation while causing targeted harm to every part of the tumour, including its vasculature. Strong antitumour immune responses that can eradicate distant metastatic lesions and residual tumour cells are characteristics of the ideal PS. Once the anticancer effect has been demonstrated, a favourable biodistribution should promote little to no off‐target effects, little or very little skin photosensitivity, and quick body clearance. These challenges to a large extent still remain active areas of concern and research in the application of photocages.
Photoactivation is an oxygen‐dependent process, which is a challenge in hypoxic sites. Furthermore, the PS utilized needs to show specificity for sites of neoplasia, and be activated by light at wavelengths that can readily penetrate into tissues. The greatest tissue penetration depth of the light employed for therapy, which is reliant on the attenuation and propagation of light in tissues, presents another difficulty for PDT treatment [177, 178]. The processes of reflectance, absorption, scattering and refraction are responsible for this reduction of light passing through tissues. The most significant factor affecting both the directionality and intensity of light attenuation in tissues, such as tumours, is the scattering of light inside such tissues. The absorption spectra of the chromophores found in tissues, including water, oxy‐ and deoxyhemoglobin, melanin and different cytochromes, establish the ideal spectrum in terms of light penetration. Absorption of light also has a significant influence on the decrease in light intensity. All of these factors which affect how light moves through different media have a major impact on the depth of penetration in tissues, and this in turn affects how well photoactivable devices can be utilised for treatment.
The “drug‐to‐light interval” (DLI) is another issue that arises while using PDT therapy [179–181]. For PDT to produce a photodynamic effect, illumination from a light source is necessary. This is carried out after the PDT medication has been administered to the body. The DLI is the amount of time that passes between the PDT drug’s administration and illumination at the appropriate wavelength. The DLI for PDT is frequently selected to cause damage to the regions of interest; notably, the tumour, the vasculature or a combination of both is dependent on the pharmacokinetics of the PS. If PS is administered intravenously and then spread to the tumour, a brief DLI would primarily target the vasculature. Given that it would have enough time to build up inside the tumour, a long DLI would primarily target the tumour cells, whereas a medium DLI would target both [182].
The tumour size may surpass the maximal penetration depth of light, making PDT less effective even when employing light inside the therapeutic window [93, 177, 183]. Consequently, the tumours that can be treated with PDT depend on the greatest propagation of the treatment light and the sites that can be reached by a light source. Because of this, PDT is frequently used to treat tumour types such as surface malignancies or conditions that may be reached with a flexible light source. These cancers include those of the bile duct, bladder, liver, colon, pancreas, brain, ovarian and prostate cancers, in addition to a number of cutaneous malignancies, such as basal cell carcinoma and squamous cell carcinoma, head and neck, esophageal and lung cancers.
The other major challenge in PDT is hypoxia [184]. The hypoxic environment that solid tumours frequently exhibit can provide a challenge to PDT’s effectiveness, since the principal acceptor of light energy, oxygen, is scarce at the site of action. This can lessen the therapeutic impact by reducing photodynamic damage through singlet oxygen production. PSs, which does not need a lot of oxygen to operate, are being used in recent attempts to alleviate hypoxia in the context of PDT [172].
15. Summary
In summary, this report discusses in depth the principles and mechanisms of both PDT and photocaging and photouncaging processes. These have been explained by the use of relevant figures and schemes for clarity. The factors that make photocaging route of drug delivery in cancer are well discussed from a background of literature reports which report laboratory scale in vitro and in vivo tests. Examples of these compounds are highlighted together with the opportunities for future research in this interesting field. Selective accumulation at sites of neoplasia (targeting) and the penetration of light as critical factors have both been discussed in the manuscript. The various biological targets and the mode of action have also been discussed. The challenges and opportunities appear in the various sections of the manuscript, with the areas of concern highlighted in the ‘challenges’ section.
16. Conclusion: Way Forward and Future Clinical Outlook
In conclusion, possible future reach and potential of light‐activatable compounds may provide real‐time drug release surveillance (i.e., real‐time radiation exposure data). For photocages, the basic method for real‐time photorelease measurement is the release or creation of a fluorescent species. Obtaining quantitative data on free drug concentration is extremely useful, especially as we move toward in vivo systems. Light‐triggered action might potentially be enhanced by imaging modalities, especially in the context of nanodevices [104].
Notwithstanding these benefits, a number of drawbacks exist when it comes to the production of these compounds. Iridium (III) complexes, for example, can be expensive and difficult to synthesize, which may prevent widespread use in medicine and large‐scale manufacturing [185]. In order to completely comprehend the long‐term effects and safety profile of these complexes, extensive in vivo toxicity studies are still necessary and pending. Furthermore, even if NIR activation enhances tissue penetration, PDT’s efficacy is still constrained by the depth of light penetration challenges, which makes it less appropriate for tumours that are deeply lodged into tissues [186].
In order to improve the bioavailability and targeting of iridium (III) complexes, future research should concentrate on creating delivery methods which impart selective distribution and regulated release of these PSs to tumour locations. These might be improved by innovations like stimuli‐responsive carriers and multifunctional nanoparticles. Examining combination treatments that combine PDT with other therapeutic approaches may improve overall therapeutic results. For example, PDT and immune checkpoint inhibitors may work in concert to enhance antitumour immune response [187]. Clinical trials and long‐term in vivo research are necessary to thoroughly assess the safety, effectiveness and any adverse effects of these complexes.
An interesting area for further study is the use of these complexes in the treatment of ailments other than cancer, such as bacterial infections and other proliferative disorders. New treatment approaches for a wider variety of medical applications might be developed through drug repurposing, by utilizing their already understood properties. In summary, although the therapeutic application of these complexes in cancer treatment shows promise, it requires the creation of effective drug delivery methods, thorough toxicity analyses and clinical protocol models, in order to successfully incorporate these complexes into clinical practice and perhaps enhance the results of cancer treatment. It is however encouraging to know that their future clinical research and the potential clinical use in cancer therapy are made possible by the ongoing developments in this sector, which are backed by strong preclinical findings.
Overall, finding innovative APIs or anticancer drug candidates remains an extremely difficult endeavour. Regulatory difficulties, distinct pharmacokinetic features of the isomers and the reliance of the photochemical properties on the physiological environment are only a few of the additional challenges that arise when one layer of complexity is added. Except for a limited number of in vivo animal experiments, cancer cell line tests are the primary means of validating biological activity at the current stage of light‐responsive anticancer medicines, which are centred on improving chemical characteristics. Due to the continual improvement of structures and rapid developments, research is moving quickly toward evaluating photoresponsive chemicals in animal models in order to validate projected advancements in cancer treatment.
Based on the foregoing discussions, the future of these photoactivable complexes will go from proof‐of‐concept PDT to a clinically feasible platform for spatiotemporally precise chemotherapy. The drawbacks, majorly on the inherent challenges of light penetration, tumour hypoxia and immune recognition, will have to be overcome by achieving localized drug activation with low systemic toxicity.
The creation of prodrugs and PSs that respond to NIR light (700–1000 nm) will be of paramount importance in the near future considering its deeper tissue accessibility, which allows for less invasive diagnosis and treatment of internal cancers. This could be made possible by coupling PSs with UCNPs, which transform NIR to visible or UV light while minimizing radiation damage to normal tissue. Recent studies have focussed on advancements in UCNP fabrication, efficiency in conversion and biological compatibility which could render this approach translationally feasible over the next 10 years.
Due to its reliance on singlet‐oxygen production, conventional PDT is constrained by tumour hypoxia. For hypoxic microenvironments, Type I PSs and PACT systems that produce radicals or superoxide by electron transfer processes are becoming attractive alternatives. Redox‐tunable chromophores and BODIPY/aza‐BODIPY composites with logical designs will increase their effectiveness in hypoxic lesions.
X‐ray or Cerenkov‐mediated PDT (X‐PDT/CR‐PDT) is another cutting‐edge method for inaccessible tumours that uses radiation technology to initiate photochemical processes deep inside tissues. At the same time, theranostic drugs that combine activation capabilities with imaging modalities like MRI, PET and fluorescence will enable real‐time therapy monitoring and planning, which is essential for clinical application. These procedures chart a viable near future application for these promising photocaged compounds.
Crosscutting support and concerted collaborations involving chemists, oncologists, immunologists and medical engineers will be necessary to realize these advancements. Within the next five to 10 years, the field of photocaged therapeutics can produce NIR‐responsive, image‐guided, immunogenic photochemotherapeutic systems. If these strategies are carefully pursued, they will be able to clinically address challenges with deep‐tissue access, with targeted tumour control.
Author Contributions
All the authors contributed similar efforts during the preparation of the manuscript.
Funding
No funding was received for this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhao Y. , Jiang X. , Liu X. , Liu X. , Liu Z. , and Liu X. , Application of Photo-Responsive Metal-Organic Framework in Cancer Therapy and Bioimaging, Frontiers in Bioengineering and Biotechnology. (2022) 10, 10.3389/fbioe.2022.1031986.PMC 963398236338113 · doi ↗ · pubmed ↗
- 2Imran M. , Ayub W. , Butler I. S. , and Zia-ur-Rehman , Photoactivated Platinum-Based Anticancer Drugs, Coordination Chemistry Reviews. (2018) 376, 405–429, 10.1016/j.ccr.2018.08.009, 2-s 2.0-85052432665. · doi ↗
- 3Yu Y. , Xu Q. , He S. et al., Recent Advances in Delivery of Photosensitive Metal-Based Drugs, Coordination Chemistry Reviews. (2019) 387, 154–179, 10.1016/j.ccr.2019.01.020, 2-s 2.0-85061790120. · doi ↗
- 4Kansız S. and Elçin Y. M. , Advanced Liposome and Polymersome-Based Drug Delivery Systems: Considerations for Physicochemical Properties, Targeting Strategies and Stimuli-Sensitive Approaches, Advances in Colloid and Interface Science. (2023) 317, 10.1016/j.cis.2023.102930.37290380 · doi ↗ · pubmed ↗
- 5Rezaei A. , Rafieian F. , Akbari-Alavijeh S. , Kharazmi M. S. , and Jafari S. M. , Release of Bioactive Compounds From Delivery Systems by Stimuli-Responsive Approaches; Triggering Factors, Mechanisms, and Applications, Advances in Colloid and Interface Science. (2022) 307, 10.1016/j.cis.2022.102728.35843031 · doi ↗ · pubmed ↗
- 6Smith N. A. and Sadler P. J. , Photoactivatable Metal Complexes: from Theory to Applications in Biotechnology and Medicine, Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. (2013) 371, no. 1995, 10.1098/rsta.2012.0519, 2-s 2.0-84887352833.PMC 368545223776303 · doi ↗ · pubmed ↗
- 7Madec H. , Figueiredo F. , Cariou K. , Roland S. , Sollogoub M. , and Gasser G. , Metal Complexes for Catalytic and Photocatalytic Reactions in Living Cells and Organisms, Chemical Science. (2023) 14, no. 3, 409–442, 10.1039/d 2sc 05672 k.36741514 PMC 9848159 · doi ↗ · pubmed ↗
- 8He M. , Chen F. , Shao D. , Weis P. , Wei Z. , and Sun W. , Photoresponsive Metallopolymer Nanoparticles for Cancer Theranostics, Biomaterials. (2021) 275, 10.1016/j.biomaterials.2021.120915.34102525 · doi ↗ · pubmed ↗