Expanding the Anatomical Distribution of PRRX1::KMT2D Fusion Mesenchymal Neoplasms: A Rare Mediastinal Case Report
Weixiang Zhong, Yu Deng, Ke Sun

TL;DR
A rare tumor with a PRRX1::KMT2D fusion was found in the mediastinum, expanding its known location and highlighting the need for advanced diagnostic methods.
Contribution
This is the first reported case of a PRRX1::KMT2D fusion tumor in the mediastinum.
Findings
The tumor was successfully resected and confirmed via pathology and RNA sequencing.
No recurrence was observed after 18 months of follow-up.
The case expands the anatomical distribution of PRRX1::KMT2D fusion neoplasms.
Abstract
PRRX1‐rearranged mesenchymal neoplasms are rare soft tissue tumors with a predilection for the superficial subcutaneous tissue. The PRRX1::KMT2D fusion variant is exceptionally rare, with only three previously reported cases, all of which were located in the intermuscular regions. However, its occurrence in deep visceral sites has not been documented. A 62‐year‐old woman was admitted after a routine physical examination revealed a space‐occupying lesion in the left thoracic cavity. Contrast‐enhanced CT showed a mixed‐density mass (10.4 × 8.1 × 3.8 cm) at the left cardiophrenic angle. The patient underwent complete thoracoscopic resection. Intraoperative frozen sections suggested a spindle cell tumor. Postoperative pathology, immunohistochemistry, and targeted RNA sequencing identified a PRRX1::KMT2D fusion mesenchymal neoplasm. At 18‐month follow‐up, no recurrence or progression was…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
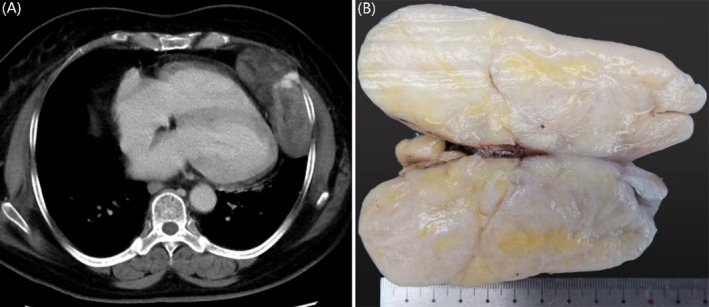 FIGURE 1
FIGURE 1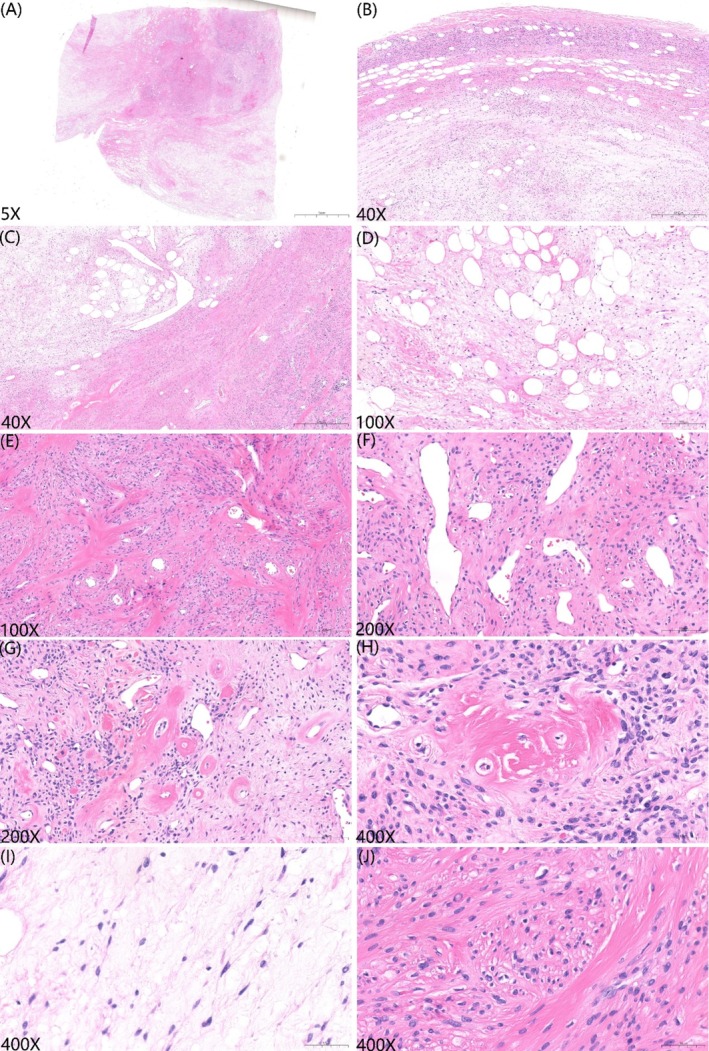 FIGURE 2
FIGURE 2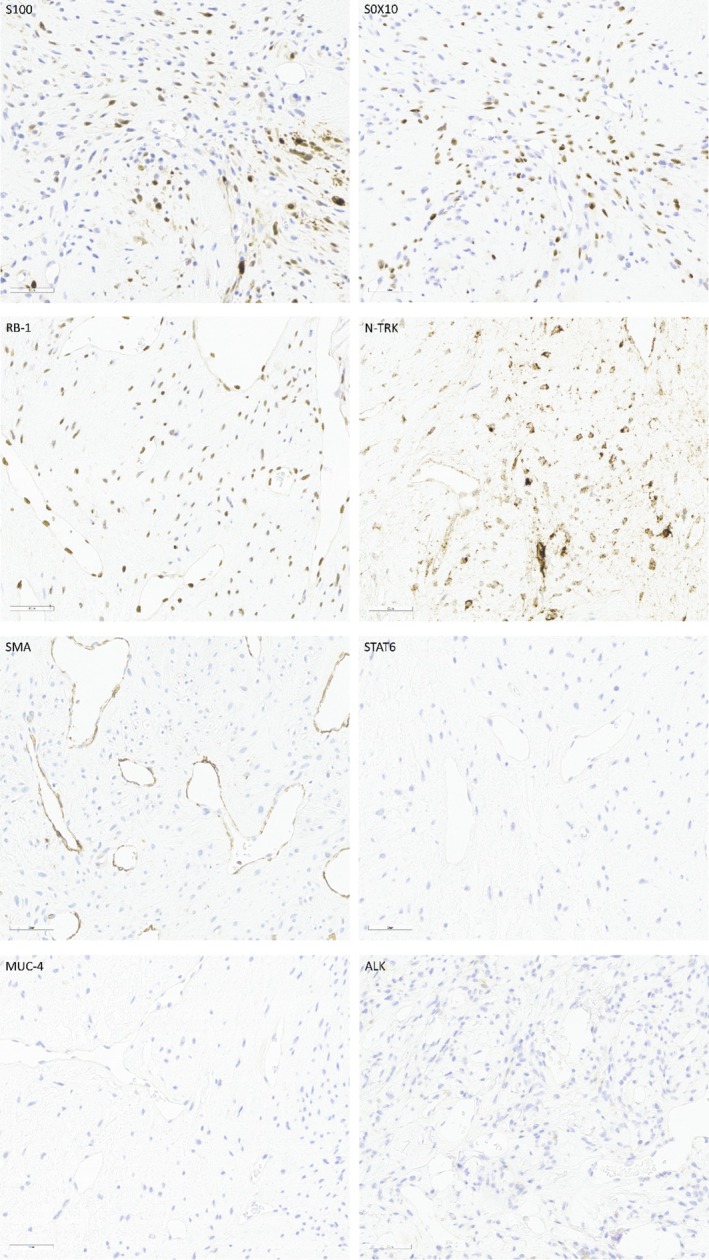 FIGURE 3
FIGURE 3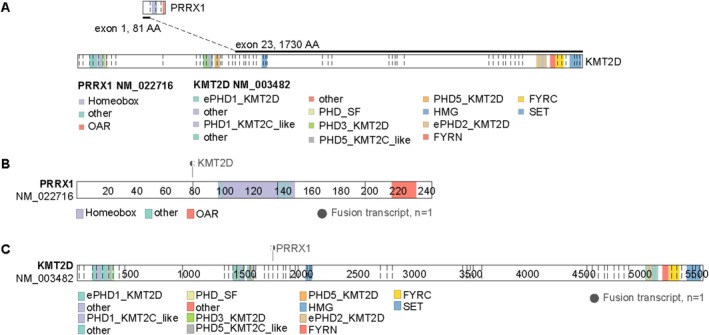 FIGURE 4
FIGURE 4| Case | Authors | Age/sex | Location | Size (cm) | S100 | SOX10 | Gene fusion | Follow‐up (months) |
|---|---|---|---|---|---|---|---|---|
| 1 | Lacambra et al. | 55/F | Thigh/S | 4 | P(weak) | NA | PRRX1ex1::NCOA1ex13 | ANED (24) |
| 2 | 33/M | Neck/S | 14 | N | NA | PRRX1ex1::NCOA1ex13 | ANED (6–18) | |
| 3 | 43/F | Neck/S | 3 | N | N | PRRX1ex1::NCOA1ex13 | ANED (6–18) | |
| 4 | 21/F | Groin/S | 2 | N | NA | PRRX1ex1::NCOA2ex15 | ANED (6–18) | |
| 5 | Puls et al. | 40/M | Knee/intramuscular | 13 | N | NA | PRRX1ex1::KMT2D ex22 | ANED (24) |
| 6 | Dermawan et al. | 49/M | Abdominal wall/S | 4 | N | NA | PRRX1ex1::NCOA1ex13 | ANED (2) |
| 7 | 43/M | Axilla/S | 5.5 | N | NA | PRRX1ex1::NCOA1ex13 | ANED (3) | |
| 8 | 34/F | Shoulder/S | N/A | N | NA | PRRX1ex1::NCOA1ex13 | N/A | |
| 9 | 41/F | Abdominal wall/S | 4 | N | N | PRRX1ex1::NCOA1ex13 | N/A | |
| 10 | 76/F | Abdominal wall/S | 2.6 | N | N | PRRX1ex1::NCOA1ex13 | ANED (2) | |
| 11 | 20/M | Hip/S | 5 | N | NA | PRRX1ex1::NCOA1ex13 | ANED (1.6) | |
| 12 | Chen et al. | 23/M | Scalp/S | 2.9 | N | N | PRRX1ex1::NCOA1ex13 | ANED (26) |
| 13 | 46/M | Groin/S | 5 | N | NA | PRRX1ex1::NCOA1ex13 | ANED (7) | |
| 14 | Cloutier et al. | 23/M | Shoulder/S | 2.5 | P(focal) | N | PRRX1ex1::NCOA1ex13 | N/A |
| 15 | Warmke et al. | 46/F | Neck/S | 2.2 | P(focal) | P(focal) | PRRX1ex1::NCOA1ex13 | ANED (4) |
| 16 | 36/M | Neck/S | 3.4 | P(focal) | P(focal) | PRRX1ex1::NCOA1ex13 | ANED (5) | |
| 17 | 65/F | Chest wall/intramuscular | 4 | P(focal) | P(focal) | PRRX1ex1::KMT2Dex25–27 | ANED (12) | |
| 18 | 37/F | Flank/S | 9.5 | N | N | PRRX1ex1::NCOA1ex13 | N/A | |
| 19 | 29/M | Forehead/frontal muscle | 2.5 | N | N | PRRX1ex1::NCOA1ex13 | N/A | |
| 20 | 56/F | Back/S | 3 | P(focal) | N | PRRX1ex1::NCOA1ex13 | ANED (16) | |
| 21 | Cheng et al. | 26/F | Left thigh/S | 4 | P(focal) | P(focal) | PRRX1ex1:: NCOA1ex15 | ANED (13) |
| 22 | Cordier et al. | 50/M | Shoulder/S | 2.7 | P | N | PRRX1ex1::NCOA1ex13 | N/A |
| 23 | 40/F | Shoulder/S | NA | N | N | PRRX1ex1::NCOA1ex13 | N/A | |
| 24 | Grosse et al. | 22/F | Supraclavicular region/S | 3 | N | N | PRRX1ex1::NCOA1ex13 | ANED (12) |
| 25 | Rongfen et al. | 40/F | Right calf/intramuscular | 15.8 | P(focal) | P(focal) | PRRX1ex1::KMT2Dex22 | ANED (1) |
| 26 | 28/F | Right thigh/S | 4 | N | N | PRRX1ex1::NCOA1ex13 | ANED (12) | |
| 27 | 26/F | Left thigh/S | 4 | N | N | PRRX1ex1:: NCOA1ex15 | ANED (14) | |
| 28 | 42/F | Flanks/S | 5.5 | P(focal) | P(focal) | PRRX1ex1:: NCOA1ex15 | ANED (3) | |
| 29 | Present case | 62/F | Mediastinum | 10.4 | P(focal) | P(focal) | PRRX1ex1::KMT2Dex23 | ANED (11) |
| Tumor type | Key diagnostic features | Molecular characteristics |
|---|---|---|
| LGFMS | “Abrupt” myxoid nodules, curvilinear blood vessels, MUC4 positive | FUS/EWSR1::CREB3L1/L2/L3/CREM fusion |
| SEF | High‐grade nuclear atypia, MUC4 positive | FUS/EWSR1::CREB3L1/L2/L3/CREM fusion |
| SFT | CD34/STAT6 positive, alternating hypercellular and hypocellular areas | NAB2‐STAT6 fusion |
| RB1‐deficient tumor family | Loss of RB1 protein, CD34/ER positive (cellular angiofibroma/mammary‐type myofibroblastoma) RB1 deletion | RB1 deletion |
| Soft tissue angiofibroma | EMA positive, variable vascular patterns | NCOA2 fusion (AHRR/GTF2I) |
| KMT2A‐YAP1 fusion sarcoma | High‐grade morphology, infiltrative growth, aggressive behavior | KMT2A fusion |
| NTRK‐rearranged tumors | Coexpression of CD34/S100, SOX10 negative, infiltrative growth | NTRK fusion |
| Nerve sheath tumors | Diffuse strong S100/SOX10 positivity (neurofibroma), EMA/claudin‐1 positive (perineurioma) | |
| Desmoid fibromatosis | Nuclear β‐catenin positivity | CTNNB1 mutation |
| OFMT | Desmin S100 CD10 and TFE3 positive | PHF1 rearrangement |
| Nasopharyngeal angiofibroma | Rare nasal tumor predominantly affecting adolescent males | CTNNB1 mutation |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSoft tissue tumor case studies · Sarcoma Diagnosis and Treatment · Gastrointestinal Tumor Research and Treatment
Introduction
1
In 2019, Lacambra et al. first described a group of mesenchymal neoplasms characterized by PRRX1 rearrangements, predominantly involving NCOA1 or NCOA2 as fusion partners [1]. Subsequent studies expanded the molecular spectrum to include KMT2D as a rare but recurrent fusion partner [2, 3, 4]. To date, only three genetically confirmed PRRX1::KMT2D fusion tumors have been reported, all situated in the intermuscular septa of the chest wall, knee, and calf muscles. Histologically, these neoplasms are characterized by hypocellular, bland spindle‐to‐stellate cells embedded within a vascularized myxocollagenous stroma [2, 3, 4].
Despite increasing recognition, the anatomical distribution and histological spectrum of PRRX1::KMT2D fusion neoplasms remain incompletely characterized. No case has been documented in visceral or intrathoracic locations prior to this report, and their distinction from histological mimics, such as NTRK‐rearranged tumors, KMT2A‐rearranged sarcomas, and RB1‐deleted neoplasms, remains diagnostically challenging [5, 6, 7, 8].
Herein, we present the first documented case of a PRRX1::KMT2D fusion mesenchymal neoplasm arising in the mediastinum. This unique presentation expands the anatomical repertoire of this emerging entity, highlights potential diagnostic pitfalls (particularly the interpretation of focal S100/SOX10/NTRK expression), and underscores the utility of targeted RNA sequencing in classifying unselected spindle cell neoplasms. By reporting this case, we aim to increase diagnostic awareness and support the inclusion of PRRX1::KMT2D fusion neoplasms in the differential diagnosis of deep‐seated myxoid spindle cell neoplasms.
Case Report
2
A 62‐year‐old woman was incidentally found to have a thoracic mass during a routine physical examination in June 2024. She had a history of mild coronary artery stenosis and reported no chest pain, dyspnea, cough, or constitutional symptoms. On June 26, the patient was admitted to the Department of Thoracic Surgery at the First Affiliated Hospital of Zhejiang University School of Medicine for further evaluation and management of the condition. Contrast‐enhanced chest computed tomography (CT) revealed a heterogeneous mass measuring 10.4 cm in size located in the left cardiophrenic angle. The lesion demonstrated several characteristic features: (1) linear and patchy areas of fat attenuation, (2) heterogeneous contrast enhancement patterns ranging from focal intensity to mild, and (3) localized posterior pleural thickening. Mild fatty liver and calcification of the aortic and coronary vessel walls were also noted. No significant lymphadenopathy or pleural effusion was observed (Figure 1A).
(A) CT image showing a heterogeneous 10.4 cm mass in the left cardiophrenic angle. (B) Gross specimen demonstrating a well‐defined, multinodular, oval mass without apparent adhesion to surrounding soft tissue. The cut surface revealed firm to translucent, with focal gelatinous areas and a pale yellow appearance.
The initial clinical impression suggested a benign lesion. After comprehensive preoperative evaluation ruled out surgical contraindications, the patient underwent complete thoracoscopic resection of the mediastinal tumor under general anesthesia on July 5, 2024. Histopathological examination confirmed negative margins. Gross examination revealed a well‐circumscribed, multinodular ovoid mass measuring 10.4 cm in greatest diameter without adhesion to adjacent soft tissues. The cut surface was variegated and composed of firm, translucent parenchyma interspersed with gelatinous areas and pale yellow discoloration (Figure 1B). Microscopic examination revealed well‐demarcated margins with subtly lobulated architecture (Figure 2A,B), featuring alternating myxoid and collagenized areas interspersed with mature adipose tissue (Figure 2C,D). The collagenous components included centrally dense coarse collagen bundles encircled by prominent irregular crescentic or “staghorn”‐patterned vascular channels displaying focal perivascular hyalinization (Figure 2E–G). At higher magnification, neoplastic cells within the myxoid regions were predominantly stellate‐shaped and presented faintly eosinophilic to clear cytoplasm and small ovoid‐to‐elongated nuclei harboring finely dispersed granular chromatin with inconspicuous nucleoli. In contrast, the collagenous zones predominantly consisted of ovoid‐spindle cells exhibiting eosinophilic cytoplasm and small ovoid‐to‐ovoid spindle nuclei, frequently demonstrating perinuclear halos and mild cytological atypia. However, mitotic activity and necrosis were absent (Figure 2I,J). Of particular diagnostic significance were the distinctive rosette‐like formations comprising neoplastic cells radially oriented around hyalinized vascular cores (Figure 2H).
Histopathological features of the specimen. (A, B) The tumor was well‐circumscribed with a thin fibrous capsule in certain regions. (C, D) The tumor consisted of myxoid and collagenized areas interspersed with mature adipose tissue. (E) Collagenized areas showed increased cellularity, arranged in loose fascicles and cords. (F, G) Irregularly dilated gaping/staghorn‐like blood vessels with prominent perivascular hyalinization. (H) Distinctive rosette‐like structures composed of increased neoplastic cells surrounding hyalinized vascular. (I, J) Tumor cells are sparse in the myxoid areas and consist of stellate‐to‐spindle‐shaped cells with small ovoid to elongated nuclei. In the collagenized areas, the cell density increased, with monomorphic ovoid to spindle‐shaped cells.
Immunohistochemical analysis revealed that tumor cells focally co‐expressing S100 and SOX10 were positive for NTRK and retained RB1 expression, while remaining consistently negative for epithelial (CKpan, EMA), myogenic (SMA), and other mesenchymal markers (CD34, STAT6, MUC4, DOG‐1, and CD117), with additional negativity for ALK. The proliferative activity, as assessed by the Ki‐67 labeling index, was low (2%) in areas with the highest degree of proliferation (Figure 3). Molecular analysis via targeted RNA sequencing (TruSight RNA Fusion Panel; Illumina, San Diego, CA, USA) identified a novel in‐frame fusion transcript between exon 1 of PRRX1 (NM_022716) and exon 23 of KMT2D (NM_003482) (Figure 4).
Immunohistochemical staining revealed that tumor cells focally co‐expressed S100 and SOX10, were positive for NTRK, and retained RB1 expression, while remaining consistently negative for epithelial markers, SMA, STAT6, MUC4, and ALK.
Targeted RNA next‐generation sequencing identified fusion transcripts of PRRX1 (NM022716) (exon 1) and KMT2D (NM003482) (exon 23). (A) Fusion occurs between exon 1 of PRRX1 and exon 23 of KMT2D, with the fusion site denoted by a bold black line and distinct colors representing the predicted domains within the protein. (B) Fusion of PRRX1 with KMT2D results in the loss of domains, such as Homeobox and OAR from PRRX1. (C) Similarly, fusion leads to the loss of domains, such as ePHD1_KMT2D, from KMT2D; whereas domains, such as HMG, remain preserved.
Based on comprehensive histopathological evaluation, immunohistochemical profiling, and molecular genetic analysis, the tumor was definitively diagnosed as a PRRX1::KMT2D rearranged mesenchymal neoplasm. The patient underwent complete surgical resection with negative margins. No adjuvant therapy was administered. Follow‐up examinations were performed every 6 months after surgery. A chest radiograph performed in January 2026 showed no evidence of tumor recurrence or metastasis.
Discussion
3
This case provides several key insights into the understanding of PRRX1::KMT2D fusion mesenchymal neoplasms. First, it documents the first mediastinal occurrence, thereby expanding the anatomical spectrum of this rare entity. Second, it confirms that deep‐seated PRRX1::KMT2D tumors can follow an indolent course similar to their superficial counterparts, in contrast to aggressive KMT2A‐rearranged sarcomas [2, 7]. Third, it highlights a diagnostic pitfall: focal S100/SOX10 co‐expression and NTRK positivity may mimic neural or NTRK‐rearranged tumors, but the absence of CD34 co‐expression and infiltrative growth, together with molecular confirmation, permit accurate classification [9]. Fourth, it underscores the utility of RNA sequencing in the evaluation of unclassified spindle cell neoplasms, particularly when immunohistochemistry yields ambiguous results.
To date, including the present case, 29 PRRX1‐rearranged mesenchymal neoplasms have been reported in the literature [1, 2, 3, 4, 5, 6, 9, 10, 11, 12]. Molecular analysis revealed NCOA1 as the predominant fusion partner (83%, 24/29), followed by KMT2D (14%, 4/29) and NCOA2 (3%, 1/29). These tumors occurred predominantly in adults (median age: 40 years; range 20–76 years), with tumor sizes ranging from 2.0 to 15.8 cm (median: 4.0 cm), and a slight female predominance (F:M = 18:11). Anatomically, most tumors (83%, 24/29) arose in superficial subcutaneous tissue, whereas 17% (5/29) originated in deep soft tissues (Table 1). Notably, the type of fusion partner appears to correlate with anatomical distribution: PRRX1::NCOAx tumors are almost exclusively subcutaneous (24/25), with only a single reported intermuscular case [3]. In contrast, all PRRX1::KMT2D‐rearranged tumors (4/4) presented deep‐seated origins: one mediastinal (present case) and three intermuscular (prior reports) [2, 3, 4]. Our report of a 10.4 cm mediastinal PRRX1::KMT2D neoplasm in a 62‐year‐old woman represents the first well‐documented occurrence outside subcutaneous or intermuscular sites.
Histopathologically, all PRRX1‐rearranged neoplasms share characteristic features, including a well‐circumscribed multinodular architecture, bland spindle cells in myxocollagenous stroma with “staghorn” vasculature, and variable additional findings (adipocytes, mast cells, and collagen rosettes). Immunohistochemically, the tumors were consistently negative for SMA, desmin, MUC4, STAT6, CD34, and EMA. Focal S100 (38%, 11/29) and SOX10 (25%, 7/20) reactivity [1, 2, 3, 4, 5, 6, 9, 10, 11, 12] suggest partial neuroectodermal differentiation [3]. This immunoprofile, particularly the concurrent S100/SOX10 expression, may mimic neural tumors, posing a potential diagnostic pitfall [3, 9]. Further challenges include RB1 deletion (FISH) and NTRK/ALK expression without molecular alterations [3, 10, 12].
The biological implications of PRRX1 and KMT2D alterations may underlie clinicopathological observations. PRRX1 (1q24.2), a paired‐related homeobox transcription factor, acts as a transcriptional coactivator that regulates morphogenesis [13]. These rearrangements are associated with therapy‐related leukemia, myelodysplastic syndromes, and musculoskeletal developmental disorders [3]. KMT2D (12q13.12, also designated MLL2) encodes a histone methyltransferase critical for differentiation and tumor suppression with mutations that are recurrent in lymphomas, carcinomas, and sarcomas [14, 15, 16]. The pathogenic role of PRRX1::KMT2D fusion in mesenchymal neoplasms requires further investigation.
Cumulative evidence supports an indolent course for PRRX1‐rearranged tumors. Accurate diagnosis is critical for distinguishing these lesions from histological mimics with malignant potential. Clinically, the differential diagnoses include thymoma, schwannoma, and teratoma. Definitive differentiation can be achieved through pathological examination. Pathological morphology overlaps with various tumors, including low‐grade fibromyxoid sarcoma (LGFMS), sclerosing epithelioid fibrosarcoma (SEF), angiofibroma, ossifying fibromyxoid tumor (OFMT), fibrosarcoma, and RB1‐deficient tumors (such as spindle cell lipoma). It is especially important to distinguish it from KMT2A‐rearranged sarcoma, which typically exhibits infiltrative growth, high‐grade morphology, and aggressive behavior [7, 8]. Although focal S100/NTRK immunoreactivity may suggest an NTRK‐rearranged tumor, the latter usually co‐expresses CD34 and is negative for SOX10 (Table 2 for a comprehensive comparison).
From a clinical perspective, recognition of this entity is critical to avoid misdiagnosis as a sarcoma and prevent overtreatment. Current evidence supports the classification of these tumors as PRRX1‐rearranged mesenchymal tumors rather than fibroblastic tumors [3]. We therefore recommend that PRRX1::KMT2D fusion neoplasms be included in the differential diagnosis of mediastinal myxoid spindle cell tumors. Definitive diagnosis relies on a systematic approach, including (1) identification of characteristic morphological features, (2) immunohistochemistry suggesting possible partial neural or neuroectodermal differentiation to exclude aggressive tissue mimics, and (3) molecular confirmation of pathogenic fusions, an algorithmic strategy particularly crucial for accurately diagnosing unclassifiable lesions encountered in limited biopsy samples.
Conclusion
4
In conclusion, we report the first mediastinal PRRX1::KMT2D fusion mesenchymal neoplasm, expanding the anatomical and morphological spectrum of this emerging entity. Our findings emphasize the importance of a multimodal diagnostic approach integrating morphological, immunohistochemical, and targeted RNA sequencing analyses. Furthermore, this case supports the indolent nature of this neoplasm. Long‐term surveillance and further case accumulation are warranted to delineate its biological and clinical characterization.
Author Contributions
All authors have made substantial intellectual contributions to this work. Weixiang Zhong was responsible for the conception of the study, data acquisition and analysis, and drafting the initial manuscript. Yu Deng participated in data interpretation and critically revised the manuscript for important intellectual content. Ke Sun supervised the project, provided final approval of the version to be published, and is the corresponding author. All authors read and approved the final manuscript.
Funding
The authors have nothing to report.
Ethics Statement
This study received ethical approval and consent for participation from the Clinical Research Ethics Board of the First Affiliated Hospital, Zhejiang University School of Medicine. The images used in this study did not contain any patient records or other identifiable information.
Consent
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. D. Lacambra , I. Weinreb , E. G. Demicco , et al., “ PRRX‐NCOA 1/2 Rearrangement Characterizes a Distinctive Fibroblastic Neoplasm,” Genes, Chromosomes & Cancer 58, no. 10 (2019): 705–712, 10.1002/gcc.22762.31008539 PMC 7104602 · doi ↗ · pubmed ↗
- 2F. Puls , A. Agaimy , U. Flucke , et al., “Recurrent Fusions Between YAP 1 and KMT 2A in Morphologically Distinct Neoplasms Within the Spectrum of Low‐Grade Fibromyxoid Sarcoma and Sclerosing Epithelioid Fibrosarcoma,” American Journal of Surgical Pathology 44, no. 5 (2020): 594–606, 10.1097/pas.0000000000001423.31913156 · doi ↗ · pubmed ↗
- 3L. M. Warmke , M. Michal , P. Martínek , et al., ““PRRX 1‐Rearranged Mesenchymal Tumors”: Expanding the Immunohistochemical Profile and Molecular Spectrum of a Recently Described Entity With the Proposed Revision of Nomenclature,” Virchows Archiv 483, no. 2 (2023): 207–214, 10.1007/s 00428-023-03575-w.37338620 · doi ↗ · pubmed ↗
- 4X. Rongfen , Z. Peipei , and W. Jian , “ PRRX 1‐Rearranged Fibroblastic Tumor: A Clinicopathological and Molecular Analysis of Four Cases,” Zhonghua Bing Li Xue Za Zhi 54, no. 4 (2025): 381–386, 10.3760/cma.j.cn 112151-20250124-00063.40159029 · doi ↗ · pubmed ↗
- 5C. Grosse , P. Noack , and A. Grosse , “ PRRX 1 ‐Rearranged Mesenchymal Tumor in a Core Needle Biopsy,” International Journal of Surgical Pathology 33, no. 6 (2025): 1454–1458, 10.1177/10668969241311492.39887195 · doi ↗ · pubmed ↗
- 6C.‐H. Chen , K.‐C. Chang , C.‐H. Chuang , J.‐T. Fu , and H.‐Y. Huang , “The Emerging PRRX 1‐NCOA Fibroblastic Neoplasm: A Combined Reappraisal of Published Tumors and Two New Cases,” Virchows Archiv 481, no. 1 (2022): 111–116, 10.1007/s 00428-021-03219-x.34647172 · doi ↗ · pubmed ↗
- 7A. Yoshida , Y. Arai , Y. Tanzawa , et al., “ KMT 2A ( MLL ) Fusions in Aggressive Sarcomas in Young Adults,” Histopathology 75, no. 4 (2019): 508–516, 10.1111/his.13926.31136005 · doi ↗ · pubmed ↗
- 8L. R. Massoth , Y. P. Hung , V. Nardi , et al., “Pan‐Sarcoma Genomic Analysis of KMT 2A Rearrangements Reveals Distinct Subtypes Defined by YAP 1–KMT 2A–YAP 1 and VIM–KMT 2A Fusions,” Modern Pathology 33, no. 11 (2020): 2307–2317, 10.1038/s 41379-020-0582-4.32461620 PMC 7581494 · doi ↗ · pubmed ↗