Genetic deletion of MrgD receptor disrupts cardiac protein homeostasis in mice
Beatriz Alexandre-Santos, Luiza Mazzali Ferraz, Ana Beatriz Proença, Nícia Pedreira Soares, Guilherme dos Santos Reis, Maria Eduarda Lima da Silva, D’Angelo Carlo Magliano, Maria Jose Campagnole-Santos, Antonio Claudio Lucas da Nóbrega, Robson Augusto Souza Santos

TL;DR
Deleting the MrgD receptor in mice leads to heart damage and protein imbalance, suggesting a new role for MrgD in heart health.
Contribution
This study reveals the novel role of MrgD receptor in maintaining cardiac protein homeostasis.
Findings
MrgD deficiency increases left ventricular mass and causes cardiac atrophy.
Genetic deletion of MrgD elevates oxidative stress and ER stress markers.
Protein degradation via ubiquitin-proteasome pathway is activated in MrgD KO mice.
Abstract
Cardiovascular diseases are the leading cause of death worldwide. An important mechanism involved is the disruption in protein homeostasis by overactivation of the classical axis of the renin-angiotensin system. The counterregulatory axis counteracts these effects; however, the MrgD receptor has recently been described, and its effects are unknown. Thus, this study aims to evaluate the impact of MrgD deficiency on cardiac protein homeostasis. 16-week-old wild-type (WT) and MrgD knockout (MrgD KO) male C57BL/6J mice were evaluated for systolic blood pressure (SBP), cardiac morphology, MDA levels, carbonyl content, and protein homeostasis markers. SBP and heart mass remained unaltered. MrgD deficiency increased left ventricular mass and led to cardiac atrophy by reduced left ventricular wall thickness and cardiomyocyte cross-sectional area. Collagen (types 1 and 3) deposition and MMP-2…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
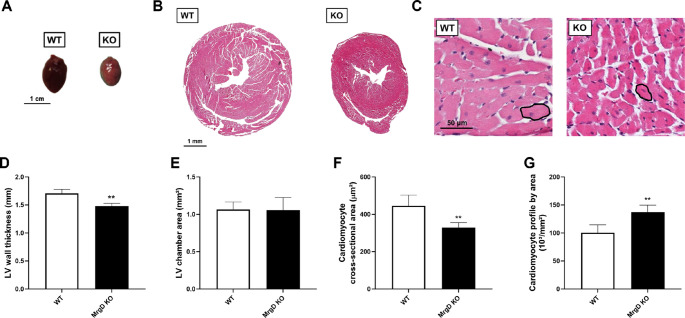 Figure 1
Figure 1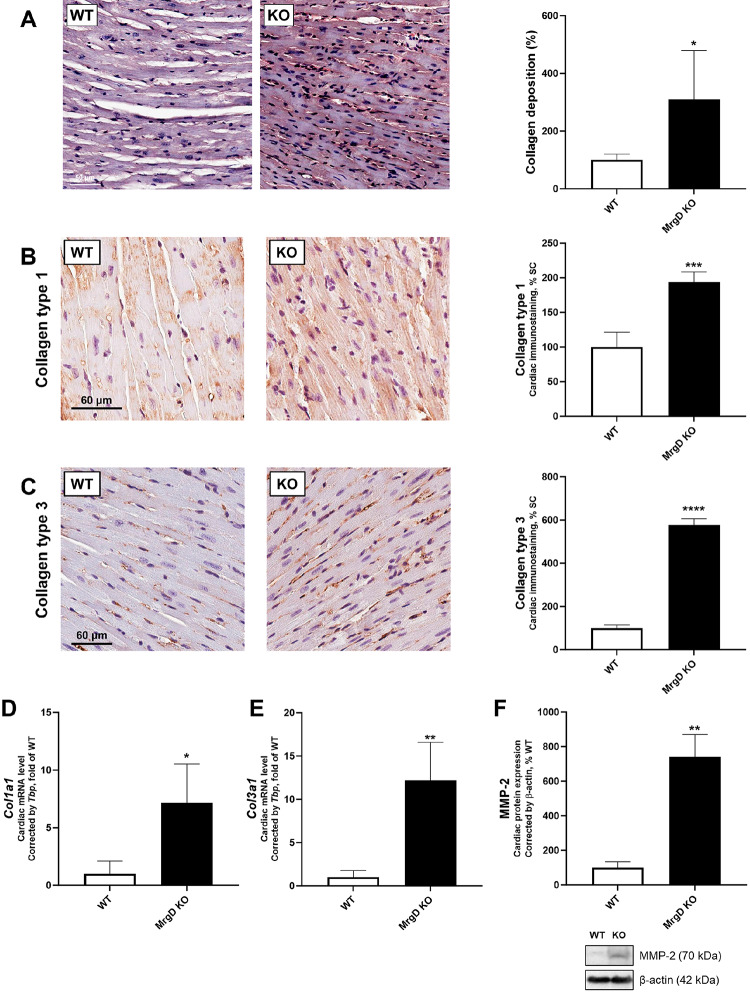 Figure 2
Figure 2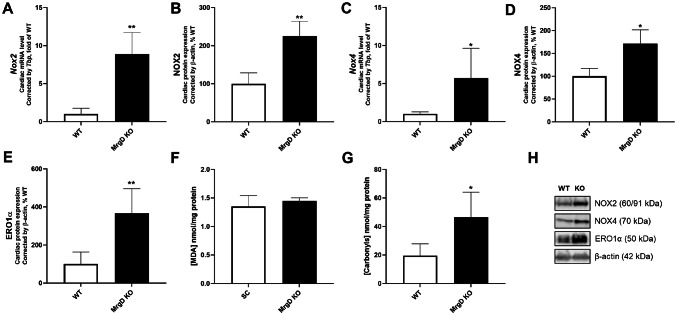 Figure 3
Figure 3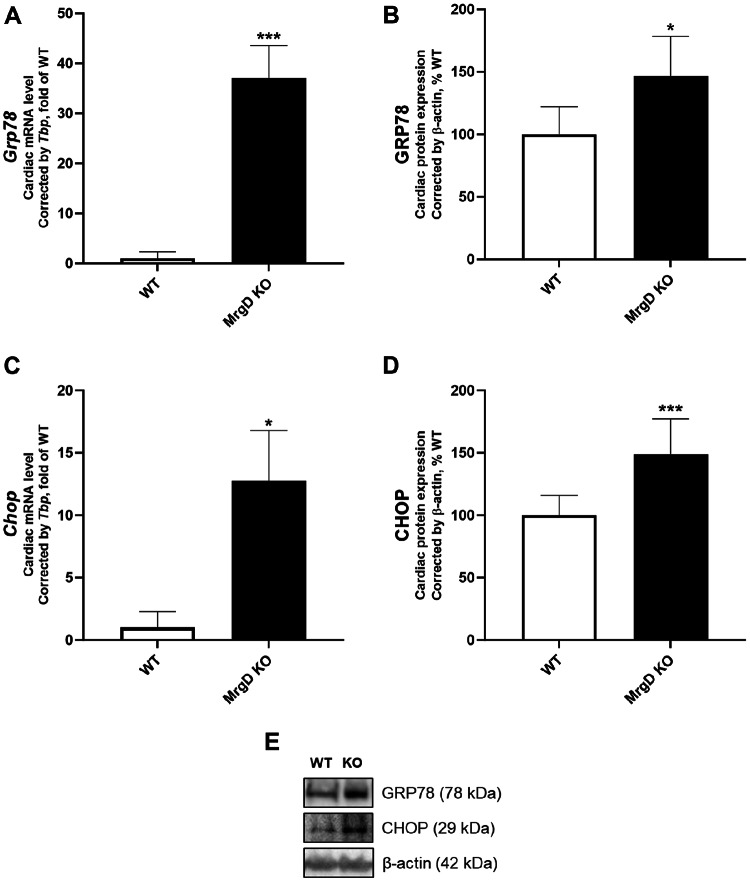 Figure 4
Figure 4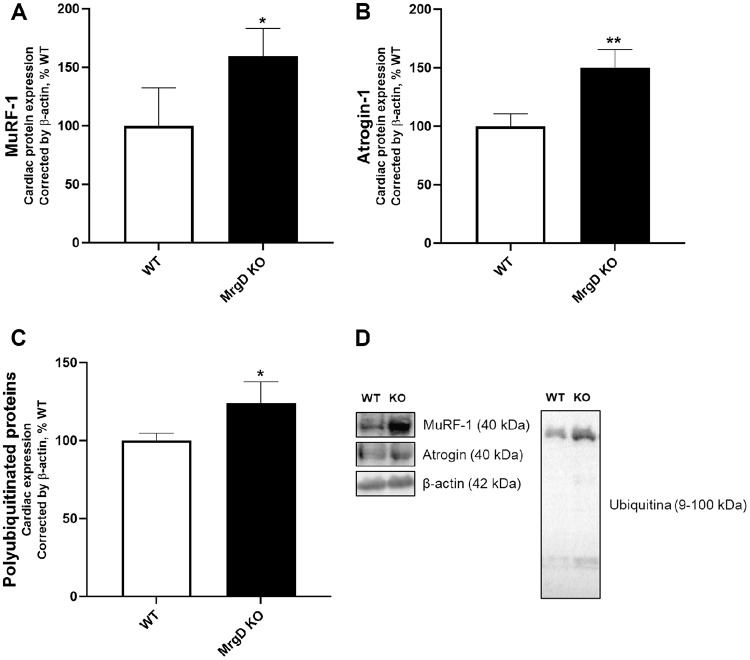 Figure 5
Figure 5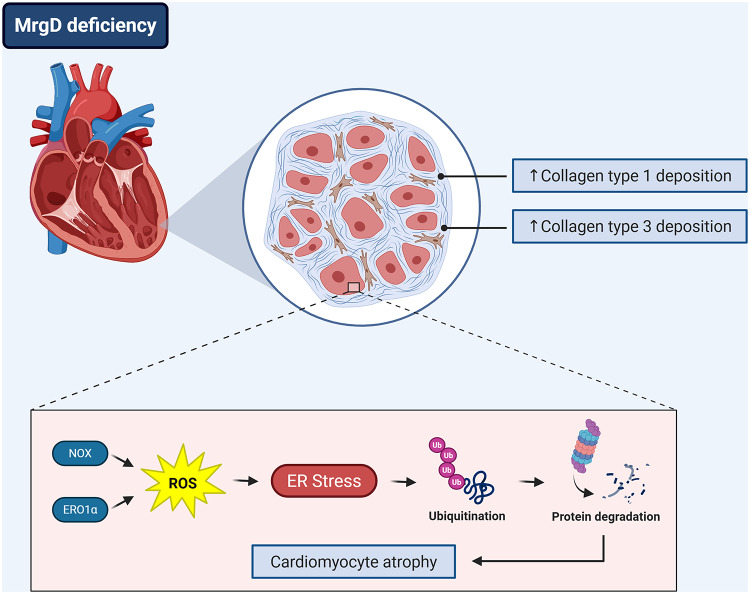 Figure 6
Figure 6- —Universidade Federal Fluminense
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital heart defects research · Cardiac Fibrosis and Remodeling · Peptidase Inhibition and Analysis
Introduction
Cardiovascular diseases (CVD) are the leading cause of death worldwide, accounting for an increasing number of deaths each year [1]. This alarming epidemiological scenario brings attention to CVD as a major global public health burden. The development and progression of CVD are multifactorial, involving a complex interplay of several pathophysiological mechanisms [2]. Overactivation of the classical axis of the renin-angiotensin system (RAS) plays a central role in the pathogenesis of several cardiovascular disorders, including hypertension, heart failure, and atherosclerosis [3, 4].
The classical axis of the RAS is composed of Angiotensin Converting Enzyme (ACE), Angiotensin (Ang)-II, and the Ang-II type 1 receptor (AT1R) [5]. When overactivated, this axis induces cardiac remodeling, encompassing hypertrophy and fibrosis, through the disruption of several cellular processes [3, 6]. These processes include excessive generation of reactive oxygen species (ROS), leading to a pro-oxidative environment that overloads cardiomyocyte protein homeostasis. Consequently, protein synthesis, folding, and quality control mechanisms within the endoplasmic reticulum (ER) are impaired [6–8]. Sustained ER stress further exacerbates proteostatic imbalance, favoring protein degradation by regulating proteolytic pathways within the ubiquitin-proteasome system [8, 9].
On the other hand, the counterregulatory axis of the RAS is traditionally composed of ACE2, Ang- [1–7], and Mas receptor (MasR) [5]. This pathway counteracts the deleterious effects of the classical axis by reducing ROS formation and attenuating disturbances in cardiac protein homeostasis [10, 11]. More recently, in 2013, a novel counterregulatory RAS pathway was identified: the alamandine/Mas-related G-protein coupled receptor type D (MrgD) pathway [12]. This pathway appears to be involved in maintaining cardiac shape and function [13, 14]. Alamandine treatment has been suggested to exert anti-inflammatory and antifibrotic effects [15], but the actions of this peptide might be mediated by other receptors or heterodimerization [14, 16]. Experimental data have demonstrated that the genetic deletion of MrgD induces dilated cardiomyopathy, underscoring its importance for cardiac integrity [17]. However, the molecular mechanisms underlying the potential cardioprotective actions of MrgD remain unexplored. Thus, this study aims to evaluate whether MrgD deficiency is involved in regulating cardiac pro-oxidative responses and protein homeostasis.
Methods
Experimental protocol
Animal care and procedures followed guidelines established in the National Institutes of Health Guide (8th edition, 2011). All experiments were approved by the Animal Ethics Committee of the Federal University of Minas Gerais (protocol number 2132023). The experimental protocol was carried out at the Transgenic Animal Facility, Hypertension Laboratory, Federal University of Minas Gerais, Brazil. Wild-type C57BL/6J (WT) and MrgD knockout (MrgD KO) mice (n = 10/group) were purchased from the Mutant Mouse Regional Resource Center (National Institutes of Health, RRID: MMRRC_036050-UNC). MrgD homozygous deficiency was validated using the following primers: MrgD-8, 5’-CATGAGATGCTCTATCCATTGGG-3’; reverse tetracycline transactivator primer (rtTA1), 5’-GGAGAAACAGTCAAAGTGCG-3’; and MrgD-1, 5’-CTGCTCATAGTCAACATTTCTGC-3’. Animals were housed in a temperature-controlled room (23 ± 2 °C) with a 12:12-h light-dark cycle, provided with food and water ad libitum.
Systolic blood pressure (SBP) was measured at 8 and 16 weeks of age by tail-cuff plethysmography (CODA, Kent Scientific, USA). At least 1 week before SBP recording, mice were acclimated for three consecutive days.
Tissue extraction
At 16 weeks old, animals were fasted for six hours, heparinized, deeply anesthetized with intraperitoneal ketamine (40 mg/kg) and xylazine (8 mg/kg), and euthanized by exsanguination. Hearts were weighed, and left ventricles (LV) were dissected, weighed, and then embedded in buffered formalin for histological analysis (n = 5 animals/group), or rapidly frozen and stored at −80°C until subsequent analysis (n = 5 animals/group).
Histological analysis
Fixed LV samples were embedded in paraffin, and 5-µm-thick sections were stained with either hematoxylin and eosin or Picrosirius Red. Digital images were obtained using the ScanScopeTM CS (Aperio Technologies, Leica, CA, USA).
In hematoxylin and eosin-stained slides, LV wall thickness and LV chamber area were assessed. Fifteen digital images, at least 10 cardiomyocytes per image, were analyzed per animal to estimate cardiomyocyte area using the digital image analysis system Image Pro Plus (version 4.5, Media Cybernetics, MD, USA). Cardiomyocyte cross-sectional area was estimated based on the numerical density of cardiomyocytes, in a frame of known area, produced by STEPanizer Web-based software [18].
In Picrosirius Red-stained slides, fifteen digital images were analyzed per animal to estimate collagen deposition. These measurements were evaluated using the digital image analysis system Image Pro Plus (version 4.5, Media Cybernetics, MD, USA) [19].
Immunohistochemistry
Fixed LV samples (n = 5/group) were embedded in paraffin, and 5 μm-thick sections were submitted to antigen retrieval and blockade (Novolink Polymer Detection Systems Kit; Leica Biosystems, RU), as described previously [20]. LV sections were incubated overnight at 4°C with primary antibodies for collagen type 1 (PAE-95137, Invitrogen) and collagen type 3 (PAD195Mu02, Cloud Clone Corp), followed by incubation with Envision FLEX/HRP (DAKO, 152 Agilent, SM802) for signal amplification. Diaminobenzidine was used for the detection of positive immunoreaction, and hematoxylin was used for counterstaining. Negative controls were performed by omitting the primary antibody. Digital images were obtained using the ScanScope CS (Aperio Technologies, CA, USA). The immunoreactivity areas were estimated using the Image-Pro Plus (Media Cybernetics, Silver Spring, MD, USA), through the density threshold selection tool. The results were presented as a percentage of the WT group [21, 22].
Western blot
Total LV proteins were extracted (n = 5/group), and protein samples were resolved by SDS-PAGE 10–15% gel electrophoresis and transferred to a polyvinylidene difluoride membrane. Membranes were incubated overnight at 4°C with primary antibodies for: matrix metalloproteinase 2 (MMP-2, PAA100Hu01, Cloud-Clone Corp), NADPH oxidase (NOX) 2 (sc-5826, Santa Cruz Biotechnology), NOX4 (sc-30141, Santa Cruz Biotechnology), endoplasmic oxidoreductin-1α (ERO1α, sc-365526, Santa Cruz Biotechnology), C/EBP-homologous protein (CHOP, sc-71136, Santa Cruz Biotechnology), 78 kDa glucose-regulated protein (GRP78, CSB-PA873500, Cusabio Biotech Co), Muscle Ring Finger protein-1 (MuRF-1, FNab08992, FineTest Biotech Inc), Atrogin-1 (PAF435Hu01, Cloud-Cone Corp), Ubiquitin (sc-166553, Santa Cruz Biotechnology). Secondary antibodies and ECL Western blot reagents were used to detect the binding of the primary antibodies. Images were acquired using the ChemiDoc system (Bio-Rad, Hercules, CA, USA). The intensity of the chemiluminescent bands was quantified using ImageJ software, version 1.44 (NIH, imagej.nih.gov/ij, USA). β-actin was used as a loading control.
Real-time reverse transcriptase polymerase chain reaction (RT-qPCR)
Total RNA was extracted from 40 mg of LV using a Trizol reagent (Invitrogen, CA, USA). RNA concentration and integrity were assessed, and synthesis of first-strand cDNA was performed. RNA samples were quantified by absorbance at 260 nm and 280 nm to determine concentration and purity. Quantitative real-time PCR was performed using an Applied Biosystems thermocycler (7500 Fast, Thermo Fisher Scientific, CA, USA) and GoTaq^®^ qPCR Master Mix (Promega, MA, USA). Constitutive gene expression (TATA box-binding protein, Tbp) was coamplified with the samples and quantified as an endogenous control. All samples were assayed in duplicate. The results were quantified by the 2 -ΔΔCt method. The primers used in this study are listed in Table S1.
Thiobarbituric acid reactive substances (TBARS) assay
Lipid peroxidation in LV homogenate (n = 5/group) was evaluated by measuring the level of thiobarbituric acid–reactive substances (TBARS) following the method of Ohkawa et al. [23]. Malondialdehyde (MDA) concentration was determined from absorbance at 532 nm and presented as MDA/mg of protein.
Protein carbonylation
Oxidative damage to proteins was measured based on the reaction between LV homogenate and dinitrophenylhydrazine (n = 5/group), as previously described [24]. Carbonyl content was determined from absorbance at 370 nm and expressed as carbonyl/mg of protein.
Statistical analysis
Data are presented as means ± standard deviation (SD). Data were tested for normality and homoscedasticity using the Shapiro-Wilk test. The differences between groups were determined by an unpaired Student’s t-test with Welch’s correction. In all cases, p < 0.05 was considered statistically significant. Analysis was performed by GraphPad Prism (version 8.0.2, La Jolla, CA, USA).
Results
MrgD deficiency induces cardiac atrophy
SBP was similar between groups at both 8 and 16 weeks (Table 1). Genetic deletion of MrgD did not affect absolute and relative heart mass but increased absolute (+ 12.35%, p < 0.05) and relative (+ 12.72%, p < 0.05) LV mass (Table 1). Cardiac size was smaller in the MrgD KO group in qualitative macroscopy (Fig. 1A). LV wall was 13.27% thinner in the MrgD KO group (p < 0.01) (Fig. 1B and D). The LV chamber area remained similar between groups (Fig. 1B and E). Cardiomyocyte cross-sectional area was smaller in the MrgD KO group (−26.14%, p < 0.01; Fig. 1C and F). Consistently, MrgD deficiency increased cardiomyocyte profile by area (+ 36.54%, p < 0.01; Fig. 1C and G).
Table 1. Biometric parametersWTMrgD KOSBP (mmHg), week 8123.7 ± 10.73127.2 ± 9.83SBP (mmHg), week 16122.3 ± 14.76122.2 ± 14.53Heart Mass (mg)134.1 ± 17.13140.4 ± 17.75Heart Mass/tibia (mg/cm)76,0 ± 10,8779,2 ± 10,11LV Mass (mg)92.3 ± 12.52103.7 ± 8.63^^LV Mass/tibia (mg/cm)51,9 ± 4,8958,5 ± 5,12^^Data presented as mean ± standard deviation (n = 10/group). Significant differences between groups are indicated with the symbols: * ≠ WT (p < 0.05), as determined by unpaired Student’s t test with Welch’s correction Fig. 1. Evaluation of cardiac morphology. (A) Macroscopic view of the whole heart (bar = 1 cm). (B, C) Representative photomicrographs of hematoxylin and eosin-stained histological sections of the heart in low (B, bar = 1 mm) and high (C, bar = 50 μm, with cross-sectional cardiomyocytes) magnifications. (D) LV wall thickness. (E) LV chamber area. (F) Cardiomyocyte cross-sectional area. (G) Cardiomyocyte profile by area. Data presented as mean ± SD (n = 5/group). Significant differences between groups are indicated with the symbols: ** ≠ WT (p < 0.01), as determined by unpaired Student’s t test with Welch’s correction
Genetic deletion of MrgD increases cardiac collagen deposition
In Picrosirius-stained slides, the MrgD KO group exhibited 209.28% higher collagen deposition (p < 0.05) (Fig. 2A). Accordingly, genetic deletion of MrgD increased collagen type 1 immunostaining (+ 93.30%, p < 0.001) and mRNA expression (+ 614.26%, p < 0.05) (Fig. 2B and D). Collagen type 3 immunostaining (+ 476.25%, p < 0.0001) and mRNA expression (+ 1121.50%, p < 0.01) were also augmented in the MrgD KO group (Fig. 2C and E). Cardiac MMP-2 protein expression was augmented in the MrgD KO group (+ 641.06%, p < 0.01; Fig. 2F).
Fig. 2. Evaluation of cardiac collagen deposition. (A) Representative photomicrographs of Picrosirius Red-stained histological sections of the heart in the same magnification (bar = 50 μm), and collagen deposition. (B) Representative photomicrographs of collagen type 1 immunostaining counterstained with hematoxylin in cardiac tissue at the same magnification in all groups (bar = 60 μm). (C) Representative photomicrographs of collagen type 3 immunostaining counterstained with hematoxylin in cardiac tissue at the same magnification in all groups (bar = 60 μm). (D) Col1a1 and (E) Col3a1 mRNA expression. (F) MMP-2 protein expression and representative Western blot analysis of proteins in LV. Data presented as mean ± SD (n = 5/group). Significant differences between groups are indicated with the symbols: * ≠ WT (p < 0.05), *** ≠ WT (p < 0.001), **** ≠ WT (p < 0.0001), as determined by unpaired Student’s t test with Welch’s correction
A pro-oxidative environment is related to MrgD deficiency
Genetic deletion of MrgD increased cardiac mRNA (+ 786.26%, p < 0.01) and protein expression (+ 125.04%, p < 0.01) of NOX2 (Fig. 3A, B and H). NOX4 mRNA (+ 470.70%, p < 0.05) and protein expression (+ 71.75%, p < 0.05) were also increased in the MrgD KO group (Fig. 3C, D and H). In addition, cardiac ERO1α protein expression was elevated in the MrgD KO group (+ 265.73%, p < 0.01) (Fig. 3E and H). Although MDA levels did not differ between groups, the MrgD KO group had higher carbonyl content (+ 139.15%, p < 0.05; Fig. 3F and G).
Fig. 3. Evaluation of pro-oxidative response. NOX2 (A) mRNA and (B) protein expression. NOX4 (C) mRNA and (D) protein expression. (E) ERO1α protein expression. (F) MDA levels. (G) Protein carbonyl content. (H) Representative Western blot analysis of proteins in LV. Data presented as mean ± SD (n = 5/group). Significant differences between groups are indicated with the symbols: * ≠ WT (p < 0.05), ** ≠ WT (p < 0.01), as determined by unpaired Student’s t test with Welch’s correction
MrgD deficiency increases ER stress and ubiquitin-related protein degradation
Regarding ER stress, cardiac GRP78 mRNA (+ 3605.90%, p < 0.001) and protein (+ 46.27%, p < 0.05) expression were higher in the MrgD KO group (Fig. 4A, B and E). Cardiac CHOP mRNA (+ 1174.30%, p < 0.001) and protein expression (+ 49.03%, p < 0.05) were also augmented in the MrgD KO group (Fig. 4C, D and E).
Fig. 4. Evaluation of ER stress. GRP78 (A) mRNA and (B) protein expression. CHOP (C) mRNA and (D) protein expression. (E) Representative Western blot analysis of proteins in LV. Data presented as mean ± SD (n = 5/group). Significant differences between groups are indicated with the symbols: * ≠ WT (p < 0.05), ** ≠ WT (p < 0.01), as determined by unpaired Student’s t test with Welch’s correction
Consistent with these findings, MrgD deficiency increased cardiac protein expression of MuRF-1 (+ 59.54%, p < 0.05) and Atrogin-1 (+ 50.18%, p < 0.01), muscle-specific E3 ligases of the ubiquitin-proteasome pathway (Fig. 5A, B and D). Moreover, the MrgD KO group had higher polyubiquitinated proteins (+ 23.94%, p < 0.05; Fig. 5C and D).
Fig. 5. Evaluation of protein homeostasis. (A) MuRF-1, (B) Atrogin-1, and (C) Polyubiquitinated proteins expression. (D) Representative Western blot analysis of proteins in LV. Data presented as mean ± SD (n = 5/group). Significant differences between groups are indicated with the symbols: * ≠ WT (p < 0.05), ** ≠ WT (p < 0.01), as determined by unpaired Student’s t test with Welch’s correction
Discussion
Genetic deletion of the MrgD receptor induced cardiac atrophy and collagen deposition, possibly due to a loss of protein homeostasis, as evidenced by the promotion of a pro-oxidative environment and ER stress. This led to the activation of the ubiquitin-proteasome pathway, a major mechanism of protein degradation (Fig. 6).
Fig. 6. MrgD deficiency disrupts cardiac protein homeostasis. MrgD deficiency led to a pattern of dilated cardiomyopathy with a remodeling process characterized by cardiac atrophy and fibrosis. Changes in cardiac extracellular matrix were evidenced by increased collagen types 1 and 3 deposition. The atrophic response was associated with increased protein degradation, which occurred as a result of a disruption in protein homeostasis. MrgD deletion increased ROS by augmenting both NOX and ERO1α protein expression, which increased ER stress and, consequently, the ubiquitin-proteasome pathway
Regarding SBP, we did not find any difference between groups. This contrasts with the previously described anti-hypertensive effects of alamandine through MrgD [12, 25, 26]. However, this is only the second study to evaluate SBP in MrgD KO animals. Oliveira et al. reported similar SBP between 12-week-old WT and MrgD KO animals, supporting our findings [17].
Consistent with the atrophic response observed herein, a previous study demonstrated, using echocardiographic evaluation, that MrgD deficiency led to dilated cardiomyopathy [17]. Although we did not observe LV chamber dilation, possibly due to a lack of hemodynamic factors, such as intracardiac pressure, in histological analysis, our findings corroborate this LV remodeling. Despite the atrophy, heart mass was not altered, and LV mass was augmented. This discrepancy between size and weight might be explained by organ histoarchitecture. Although smaller, the hearts of MrgD KO animals had increased cardiac collagen expression and deposition, and collagen is denser than normal interstitium. This is the first study to directly assess heart weight and extracellular matrix features in MrgD KO animals. However, Oliveira et al. found, by echocardiography, that genetic deletion of MrgD did not alter ventricular mass in 2-week-old mice but increased this parameter in 12-week-old mice [17]. The pro-fibrotic effect of MrgD deficiency is consistent with the reported anti-fibrotic effect of alamandine. In different models of myocardial infarction and hypertension, alamandine treatment has been shown to reduce collagen deposition and MMP-2 and transforming growth factor-β (TGF-β) expression [27–29]. Although our findings suggest a pattern of structural remodeling that is commonly associated with cardiac dysfunction and increased myocardial stiffness, the lack of functional analysis limits the confirmation of dilated cardiomyopathy as previously described [17]. Despite that, similar morphological responses were observed, corroborating the intimate relation played by function and morphology in cardiac pathophysiology [30].
This pronounced LV remodeling upon MrgD deficiency was accompanied by a pro-oxidative environment, with increased ROS formation resulting from both superoxide production by NOX and hydrogen peroxide by ERO1α. Oxidative damage may result from lipid peroxidation, protein oxidation, and DNA oxidation [31]. Genetic deletion of MrgD augmented protein oxidation, but lipid peroxidation remained unaltered. Interestingly, a pro-oxidative response is known to contribute to the sustained accumulation of unfolded and misfolded proteins, thus triggering the unfolded protein response (UPR) and ER stress [8]. Indeed, ER stress markers were increased in MrgD KO animals. This is the first study to report any impact of MrgD on ER stress, and studies addressing other counterregulatory components remain scarce. In agreement with our findings, few data available suggest that reducing the Ang- [1–7]/MasR pathway leads to UPR activation [8], indicating an involvement of the counterregulatory axis in ER stress regulation.
The UPR is a key regulator of proteolytic pathways, particularly the ubiquitin-proteasome system, aiming to reestablish protein homeostasis [32]. In the heart, this process depends on muscle-specific E3 ligases, i.e., MuRF-1 and Atrogin-1, which are hallmarks of muscle atrophy [33–35]. These markers were increased in MrgD KO animals, which suggests activation of the ubiquitin-proteasome pathway. Corroborating these findings, MrgD deficiency increased polyubiquitinated proteins. The role of the counterregulatory axis in muscle atrophy is poorly explored. In skeletal muscle, the Ang- [1–7]/MasR pathway appears to have a protective role, reducing MuRF-1 and Atrogin-1, and preventing protein polyubiquitination [36, 37]. To date, this is the first study to associate MrgD with markers of protein degradation.
Although this study only evaluated male mice, the previous study characterizing dilated cardiomyopathy in MrgD-deficient mice observed that this effect is independent of sex [17]. Thus, this does not blunt the relevance of the findings described herein.
Conclusions
Taken together, our findings demonstrate that the cardiac effects of genetic deletion of MrgD are associated with disturbances in protein homeostasis, characterized by an increase in protein degradation. This may contribute to the structural alterations observed, with cardiac atrophy and fibrosis. Importantly, these results further reinforce the protective role of MrgD in maintaining cardiac integrity, suggesting that its presence is essential for preserving protein stability.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bader M, Muscha Steckelings U, Alenina N, Santos RAS, Ferrario CM (2024) Alternative Renin-Angiotensin System. Hypertens (Dallas, Tex 1979) https://pubmed.ncbi.nlm.nih.gov/38362781/10.1161/HYPERTENSIONAHA.123.21364 PMC 1102380638362781 · doi ↗ · pubmed ↗
- 2Azevedo PS, Polegato BF, Minicucci MF, Paiva SAR, Zornoff LAM (2016) Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arquivos brasileiros de cardiologia 106:62–6910.5935/abc.20160005 PMC 472859726647721 · doi ↗ · pubmed ↗
- 3Jesus ICG, Mesquita T, Souza Santos RA, Guatimosim S (2023) An overview of alamadine/Mrg D signaling and its role in cardiomyocytes. Am J Physiol Cell Physiol 324(3):C 606-C 613. https://pubmed.ncbi.nlm.nih.gov/36571443/10.1152/ajpcell.00399.2021 PMC 1103369436571443 · doi ↗ · pubmed ↗
- 4Oliveira AC, Melo MB, Motta-Santos D, Peluso AA, Souza-Neto F, Da Silva RF et al Genetic deletion of the alamandine receptor MRGD leads to dilated cardiomyopathy in mice. Am J Physiol Heart Circ Physiol [Internet]. 2019 Jan 1 [cited 2024 Mar 27];316(1):H 123–33. Available from: https://pubmed.ncbi.nlm.nih.gov/30339496/10.1152/ajpheart.00075.201830339496 · doi ↗ · pubmed ↗
- 5Barbosa-da-Silva S, Fraulob-Aquino JC, Lopes JR, Mandarim-de-Lacerda CA, Aguila MB (2012) Weight cycling enhances adipose tissue inflammatory responses in male mice. P Lo S One [Internet]. Jul 25 [cited 2025 Jul 3];7(7). Available from: https://pubmed.ncbi.nlm.nih.gov/22848362/10.1371/journal.pone.0039837 PMC 340508622848362 · doi ↗ · pubmed ↗
- 6Mandarim-de-Lacerda CA, Fernandes-Santos C, Aguila MB (2010) Image analysis and quantitative morphology. Methods Mol Biol 611:211-225. https://link.springer.com/protocol/10.1007/978-1-60327-345-9_1710.1007/978-1-60327-345-9_1719960334 · doi ↗ · pubmed ↗
- 7Gella A, Durany N (2009 Jan-Mar) Oxidative stress in Alzheimer disease. Cell Adh Migr 3(1):88–93. https://www.tandfonline.com/action/journal Information?journal Code=kcam 2010.4161/cam.3.1.7402 PMC 267515419372765 · doi ↗ · pubmed ↗
- 8Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA (2008) The initiation factor e IF 3-f is a major target for atrogin 1/MA Fbx function in skeletal muscle atrophy. EMBO J 27(8):1266-1276 https://www.embopress.org/doi/pdf/10.1038/emboj.2008.52?download=true 10.1038/emboj.2008.52PMC 236739718354498 · doi ↗ · pubmed ↗