Contribution of tumor-derived extracellular vesicles in the establishment of the pre-metastatic niche: lessons learned from past experimentations and future directions
Laurence Blavier, Andjela Crnjac, Yves A. DeClerck

TL;DR
This paper reviews how tumor-derived extracellular vesicles help create environments in the body that support cancer spread, and suggests future research directions.
Contribution
The paper systematically reviews 120 studies to identify seven functional roles of tumor-derived extracellular vesicles in metastasis and proposes future experimental approaches.
Findings
Tumor-derived extracellular vesicles contribute to the pre-metastatic niche through seven functional hallmarks like immune reprogramming and ECM remodeling.
Exogenous and endogenous models of extracellular vesicles each have strengths and limitations in studying metastasis.
Loss-of-function experiments are needed to determine if targeting these vesicles can prevent cancer metastasis.
Abstract
Tumor-derived extracellular vesicles (TEVs) have been shown to actively contribute to the establishment of the pre-metastatic niche (PMN) through multiple mechanisms of action and the transfer of cargo material to host cells. Here, we report a review of 120 manuscripts published between January 2010 and April 2025 describing observations from in vivo experiments aimed at the examination of the contribution of TEVs to the PMN and the metastatic niche (MN). Whereas most of these publications reported observations made with models of exogenous administration of TEVs prepared in vitro, five publications used endogenous models of TEVs released by implanted tumors to track their fate and examine their role in the PMN. Breast and colon cancers and melanoma are the most common types studied, and lung, liver, bone, and lymph nodes are the most common sites of metastasis examined. Methods to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
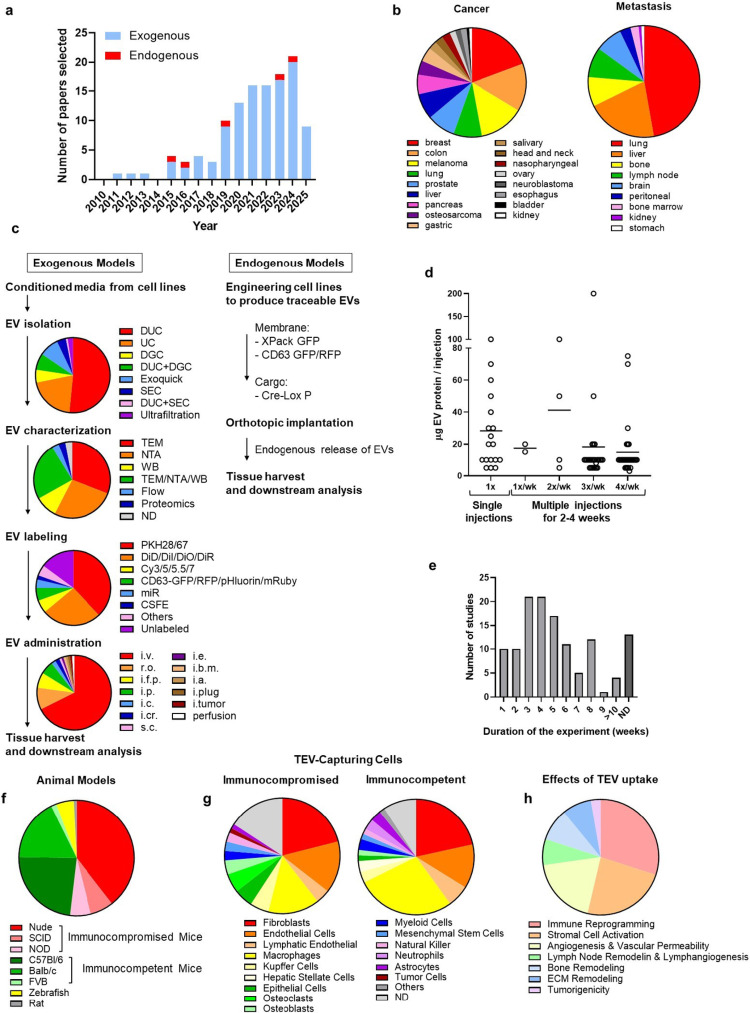 Figure 1
Figure 1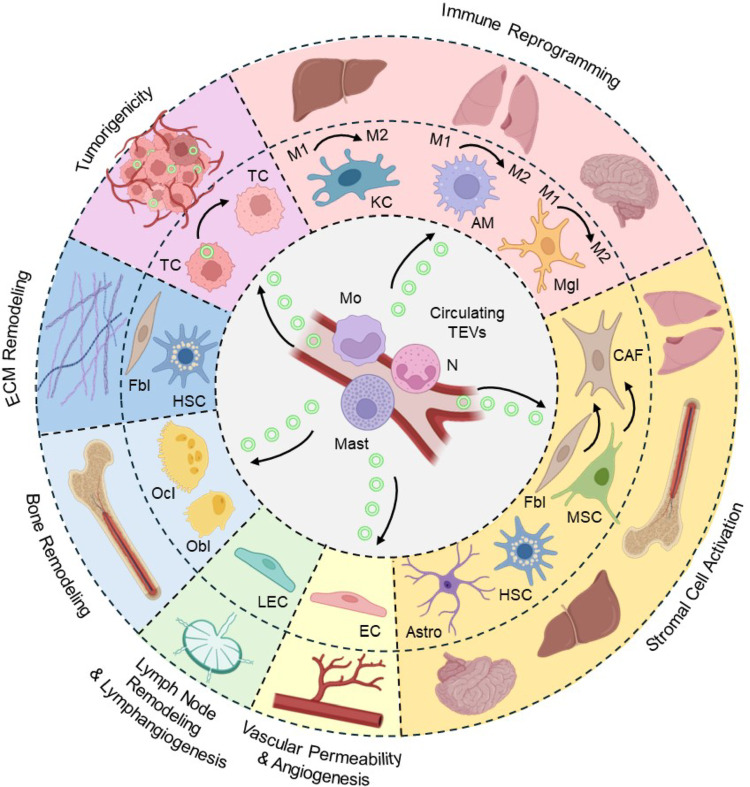 Figure 2
Figure 2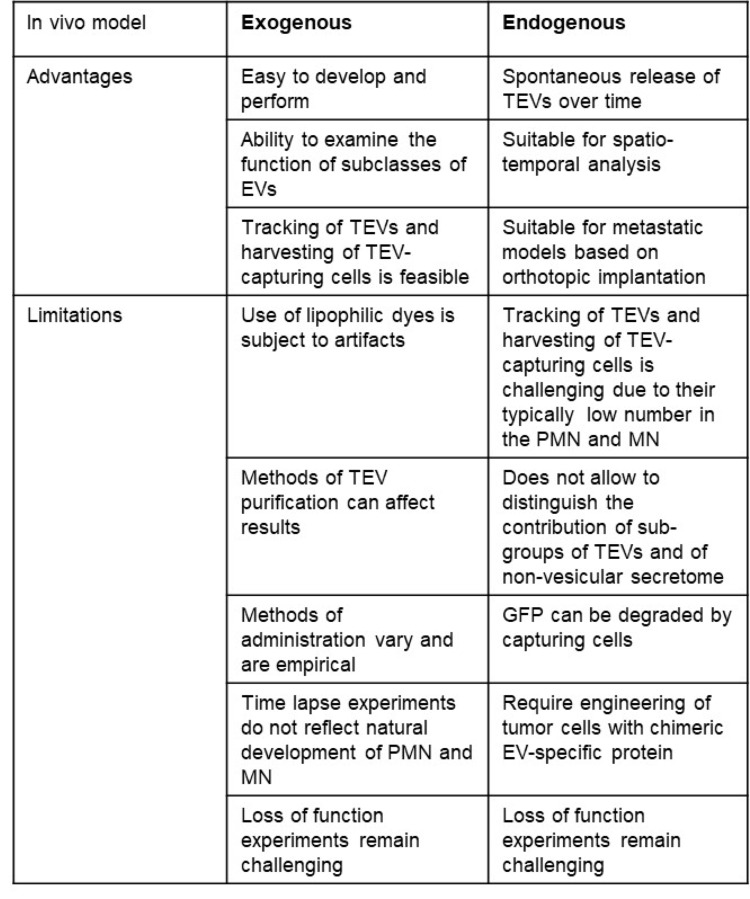 Figure 3
Figure 3- —University of Southern California
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsExtracellular vesicles in disease · Nanoplatforms for cancer theranostics · Cancer Cells and Metastasis
Introduction
Metastasis is a major cause of failure to treat cancer and despite significant advances in our understanding of its mechanisms it remains difficult to treat or to prevent. Considerable progress has been made in elucidating the mechanisms of metastasis, since 1889 when Stephen Paget proposed the “Seed and Soil” theory [1]. The validity of this theory was experimentally confirmed by Isaiah Fidler who -using the B16 murine melanoma model- demonstrated the importance of the organotropism of tumor cells in determining their metastatic behavior [2]. For long, the accepted scientific concept has been that tumor cells have the ability to colonize every organ but are only able to proliferate in organs providing a microenvironment that promotes angiogenesis, tumor cell proliferation and immune escape [3]. Experiments in many cancers demonstrated that metastatic tumor cells have the potential to reprogram the microenvironment at the site of metastasis to their advantage, a process described as the formation of the metastatic niche (MN) [4, 5]. However, in 2006 R. Kaplan and colleagues at Cornell University, using murine models of metastatic melanoma and lung carcinoma, challenged this concept by providing experimental evidence that bone marrow-derived cells (BMDC) colonize organs prone to metastasis prior to the homing of metastatic tumor cells. Thus, alterations in the microenvironment at metastatic sites can occur before and not after metastatic tumor cells arrive, a process described as the formation of the pre-metastatic niche (PMN) [6]. The establishment of the PMN is a complex, dynamic, and evolving process [7]. In its early stage, it is characterized by extracellular matrix (ECM) remodeling and inflammatory changes caused by tissue-resident immune and stromal cells. This stage is followed by an enhanced deposition of ECM proteins such as fibronectin and tenascin C, and an increase in vascular permeability that facilitates the recruitment of BMDC and later tumor cells. In its latest stage, the PMN is characterized by an infiltration of immunosuppressive myeloid cells (neutrophils and monocytes), tumor-associated macrophages (TAM) and endothelial progenitor cells that contribute to the formation of a myeloid-rich, lymphoid-poor immunosuppressive and pro-angiogenic microenvironment [8–11]. In a prospective study in patients with pancreatic cancer who had liver biopsy done at the time of surgical tumor resection, investigators provided indirect evidence for the formation of a PMN in patients with cancer. They reported that the liver of patients who later developed metastasis exhibited signs of augmented inflammation with an enrichment of neutrophil extracellular traps (NETs), Ki-67 upregulation and decreased liver creatine that significantly distinguished them from livers of patients who did not developed metastasis and the livers of non-cancer control patients [12].
The existence of a PMN raised the central question of the mechanisms by which tumor cells distantly communicate with cells in the future PMN to induce changes favorable for their implantation. The demonstration that the organotropism of metastatic tumor cells in vivo can be altered upon priming with the conditioned medium of tumor cells brought the attention to soluble factors including growth factors and cytokines [13] and extracellular vesicles (EVs) [14]. These latter, represent a heterogeneous group of small organelles with a lipid-bilayer membrane ranging in size from 50 nm to > 10 μm in diameter, and designated according to their biogenesis and size as exosomes (< 200 nm), ectosomes (200–400 nm), migrasomes (> 500 nm), apoptotic bodies (> 1 μm), or oncosomes (10 μm) [15, 16]. EVs are released by all living cells and serve as important vehicles in cell-to-cell communication. A unique characteristic of EVs is their cargo rich in a large variety of proteins, lipids, metabolites, and nucleic acids, reflecting the cells from which they originate. Methods to isolate, purify and characterize EVs are numerous, some more rigorous than others and they vary in the type of EVs they isolate [17].
There is abundant evidence that tumor-derived EVs (TEVs) represent one of the endocrine mechanisms adopted by tumor cells to trigger the formation of the PMN and the recruitment of immuno-suppressive BMDC to the PMN. However, such evidence has been hampered by technical limitations in the ability to track TEVs and document their capture in the PMN, and by the animal models used. In this article we performed a systematic review of fifteen years (2010–2025) of peer-reviewed scientific publications reporting the role of TEVs in metastasis and the establishment of the PMN in animal models. We identified 120 publications which were reviewed for the methods reported to purify, characterize, administer and track TEVs in vivo. Cancer types, metastatic sites, cells capturing TEVs and effects of TEV-capture on the PMN and/or MN were recorded and tabulated. Observations made on exogenous and endogenous models were summarized and the article ends by identifying the future challenges in targeting TEVs to inhibit or prevent metastasis in patients with cancer.
Methods
Search and selection of publications
Publications were selected using PubMed Advanced Search Builder with the following search queries: “(“extracellular vesicles“[Title/Abstract] OR “EV“[Title/Abstract] OR “exosomes“[Title/Abstract] OR “endogenous release“[Title/Abstract]) AND (“metastatic“[Title/Abstract] OR “metastasis“[Title/Abstract] OR “pre-metastatic“[Title/Abstract] OR “premetastatic“[Title/Abstract] OR “pre-metastasis“[Title/Abstract]) AND (“metastatic niche“[Title/Abstract] OR “pre-metastatic niche“[Title/Abstract] OR “premetastatic niche“[Title/Abstract] OR “MN“[Title/Abstract] or “PMN“[Title/Abstract]) NOT (Review[Publication Type])”. The range of years selected was January 1, 2010-April 15, 2025. The list obtained was further sorted by removing additional review papers, papers reporting a role for non-tumor derived EVs and papers reporting work solely done in vitro. Publications were organized in an excel spreadsheet format and chronologically organized by PMID (Supplemental Table 1). Three publications that did not contain in their title or abstract the key words used in our search, were however included in our list of 120 papers because of their relevance [14, 18, 19].
Publication analysis
Each publication was examined for the following aspects: (1) Type of cancer and metastatic site(s) studied, (2) Method(s) to isolate TEVs in exogenous models, (3) Methods to characterize TEVs, (4) Methods to label and track TEVs in vivo, (5) Models of TEV administration/release, (6) Amount of TEVs administered, (7) Duration of the experiment, (8) Metastatic model (immunocompetent vs. immunodeficient, experimental vs. orthotopic model), (9) Cells capturing TEVs, and (10) Effect of TEVs on the PMN or MN. The data were recorded and sorted using Microsoft Excel data collection and graphed using GraphPad Prism and BioRender.
Results
Exogenous models
Our research identified a total of 120 peer reviewed original publications reporting a contribution of TEVs to metastasis between 2010 and 2025 with a significant increase after 2018. Among those, 115 used an exogenous model (Fig. 1a and Supplemental Table 2). These publications cover a large variety of cancer types with a majority being breast and colon cancers, and melanoma. Among the metastatic organs studied, lung, liver, bone, and lymph nodes were the most commonly examined with few reports on bone marrow, brain and kidney metastasis (Fig. 1b and Supplemental Table 3). The methods used in these models and the effects observed in the PMN and MN are summarized below. In exogenous models, TEVs were isolated, characterized, labeled, and administered using a large variety of methods (Fig. 1c). Differential ultracentrifugation (DUC) was the most common method used (56% of the reports) followed by ultracentrifugation (UC) (17% of the reports). OptiPrep™ density gradient centrifugation (ODGC) or sucrose density gradient centrifugation were used in 11% and size exclusion chromatography (SEC) in 4%. In 8% of the reports, a combination of two techniques (UC or DUC and SEC in 0.9% or UC/DUC and density gradient in 7%) was reported. Precipitation methods like ExoQuick^®^, were used in 9%. As a result, some of these publications refer to exosome whereas others, and particularly the most recent use the term EV. Among the 115 publications using exogenous models, 95% provided a detailed characterization of the material collected including transmission electron microscopy (TEM), or nano tracking analysis (NTA) to determine the size of the particles, and Western Blot (WB) to demonstrate the presence of proteins typically associated with EVs such as tetraspanins. A combination of these three methods was used in 35% of the papers, mostly in the most recent years in compliance with updated guidelines from the International Society of Extracellular Vesicles [17]. In 81% of the cases, TEV labeling was used as a second step in the
Fig. 1a Research papers published over the last fifteen years describing a role of TEVs in the pre-metastatic niche. b Pie diagrams representing the proportion of papers published per cancer models studied (left) and the proportion of papers addressing the listed metastatic sites (right). c Comparison between methods used for exogenous administration of TEVs versus the methods used in models of endogenous release of TEVs. Diagrams represent the proportion of papers using the listed techniques of TEV isolation, characterization, labeling, and administration. d Comparison of doses of TEV protein used in single and multiple administrations. e Duration of the experiment to analyze PMN and MN. f Proportions of studies done using different animal models, including immunocompromised vs. immunocompetent mice general backgrounds. g Proportions of studies identifying the listed TEV-capturing cells. h Proportions of studies identifying the listed hallmark effects of TEV-capture on the PMN and MN. DUC, differential ultracentrifugation; UC, ultracentrifugation; DGC, density gradient centrifugation; SEC, size exclusion chromatography; TEM, transmission electron microscopy; NTA, nano tracking analysis; WB, western blot; i.v., intravenous; r.o., retroorbital; i.f.p., intra food pad; i.p., intraperitoneal; i.c., intracardiac; i.cr., intracranial; s.c., subcutaneous; i.e., intra ear; i.b.m., intra bone marrow; i.a., intra anal
preparation of TEVs to allow their tracking in vivo. Lipophilic fluorescent dyes like Paul Karl Horan (PKH, 36 %) dyes, carbocyanine dyes (DiD, Dil, DiO, or DiR, 24 %), or near infra-red fluorescent dyes Cy5.5 or Cy7 (5 %) were commonly used. In 62 % of those cases, TEV labeling was followed by density gradient centrifugation to remove excess dye. The routes of exogenous administration of TEVs differ among the publications reviewed. Intravascular systemic administrations were common routes (77.1 % intravenous and 1.9 % intraarterial), whereas local administration was used in 21 % of the studies. Doses and frequency of TEV administration widely ranged from 5-100 μg/mouse of TEV proteins in a single administration to 5-200μg/mouse of TEV proteins given 1-4 times per week over 2-4 weeks (Fig. 1d). Single injections (30%) were typically used to assess biodistribution of the TEVs, whereas multiple administrations (70%) were used to educate the PMN prior to the administration of cancer cells. In the five publications that used endogenous release of TEVs, tumor cells were engineered with a chimeric luminescent (luciferase) or fluorescent protein (GFP or RFP-tetraspanin CD63 or GFP-link kinase (XPack)) and the tracking of labeled TEVs was performed in animals implanted orthotopically. In all papers examined, the duration of the experiment required to analyze the PMN varied greatly, from one up to more than 10 weeks (Fig. 1e). A murine model was used in 94.1% of the studies with 55.4 % being done in immunodeficient and 44.6% in immunocompetent mice. Six reports used a Zebrafish model for its convenient transparency ideal for the tracking of TEVs and tumor cells. One publication reported a study done on rats (Fig. 1f). The different cell types capturing TEVs were similar in immunocompromised and immunocompetent animals, with the majority being identified as macrophages, fibroblasts, and endothelial cells (EC) (Fig. 1g).
The organotropism of TEVs is directed by adhesion molecules and TEV-capturing cells are organ specific
In a seminal paper, Hoshino et al. used PKH and near infra-red-labeled TEVs from MDA-MB-231 human breast cancer cells and subclones with organotropism for bone, lung, and brain, as well as TEVs from colorectal and pancreatic cancer cells metastatic to the liver, and demonstrated that TEVs have specific organotropism for these organs that is controlled by integrins with TEVs containing integrins α_6_β_4_ and α_6_β_1_ seeding in the lung and being associated with lung metastasis, while TEVs containing integrin α_v_β_5_ being retained in the liver and associated with liver metastasis. These TEVs are captured by resident cells mainly Kupffer cells (KC) in the liver, EC in the brain and fibroblasts and epithelial cells in the lung [20]. Another study has shown that TEVs from urological cancer cells injected intravenously are taken up within 12 h by the lung, liver, brain and spleen but not by other organs and that the level of incorporation in these organs depends on the type of cancer, with the lung being the dominant organ in prostate and bladder cancers and the brain in kidney cancers [21]. In breast cancer, the expression of the melanoma cell adhesion molecule (MCAM/CD146/MUC18) in TEVs allows them to be targeted to the lung [22].
Seven hallmarks of TEVs
The uptake of TEVs has a broad spectrum of effects on the PMN and MN that is a function of the cell type and the organ being targeted (Fig. 1h). These effects are summarized here in seven functional categories, designated hallmarks (Fig. 2): (1) Immune reprogramming, (2) Stromal cell activation, (3) Vascular permeability and angiogenesis, (4) Lymphatic remodeling and lymphangiogenesis, (5) Bone remodeling, (6) ECM remodeling and (7) Direct tumorigenicity.
TEVs reprogram immune cells
TEVs are captured by immune innate cells like macrophages, monocytes, neutrophils, and mast cells which results in the reprogramming of their immune function often but not always toward an immune suppressive role. TEVs are preferentially taken by resident macrophages but also bone marrow-derived and circulating monocytes. Macrophages from the liver and KC capture TEVs from colorectal cancer (CRC) cells and gastric cancer cells which polarize them into M2 macrophages promoting metastasis [23–26]. TEVs containing angiopoietin like protein-1 taken by KC in the liver decrease the secretion of matrix metalloproteinase (MMP)-9 and improve vascular integrity [27]. In the lung, TEVs promote an immunosuppressive activity in macrophages by upregulating their expression of program death ligand (PDL)-1 resulting in T cells suppression and exhaustion [28, 29]. Such M2 polarization occurs through activation of several signaling pathways like ERK1/2, PI3K/AKT/mTOR, STAT3, YAP and TLR-NFkB [25, 30–33]. As a result, polarized macrophages secrete cytokines and chemokines like CCL2 [34], CXCL1 [35], IL-6 [36] and IL-8 [37, 38] attracting innate immune myeloid cells to the PMN such as myeloid derived suppressive cells (MDSC) and neutrophils [39, 40]. TEV-capturing macrophages (and lymphatic EC) can also act as tumor antigen-presenting cells inducing T cell apoptosis [41]. In the brain, microglial cells capture TEVs which promote their M2 polarization by regulating a miR-142p /TGFβ pathway that facilitates brain metastasis [42]. Peritoneal macrophages capturing TEVs play a role in the dissemination of ovarian and gastric cancers as they polarize toward M2 and secrete CCL2, CXCL5 and CCL5 [43, 44]. Bone marrow-derived monocytes capture TEVs in the bone marrow and the blood circulation and mature into M2 macrophages [45, 46]. However, some, like Ly6C^low^ patrolling monocytes, can have an inhibitory effect on metastasis as they capture TEVs from low metastatic cells and inhibit the growth of highly metastatic cells [47, 48]. A particular aspect of macrophages is their ability to transfer TEVs they have captured to other cells. They transfer TEVs to EC promoting angiogenesis [49] and to fibroblasts [50] stimulating tumor cell invasion. In immunodeficient mice, TEVs from osteosarcoma cells promote the recruitment of CD11b myeloid cells to the lung however without forming a functional PMN [51]. Mast cells, which play a promoting role in melanoma metastasis, capture the DNA-binding protein high mobility group AT-hook 1 (HMGA1) secreted by melanoma cells which contribute to their pro-metastatic activity [52]. Like macrophages, neutrophils capture circulating TEVs in the lung which activates TLR/NFkB signaling, the production of IL-1β [53] and their N2 polarization [54].
Fig. 2. The seven hallmark effects of TEV-uptake on PMN and MN. KC, kupffer cell; AM, alveolar macrophage; Mgl, microglial cell; CAF, cancer associated fibroblast; Fbl, fibroblast; MSC, mesenchymal stromal cell; HSC, hepatic stellate cell; Astro, astrocyte; EC, endothelial cell; LEC, lymphatic endothelial cell; Obl, osteoblast; Ocl, osteoclast; TC, tumor cell; M, monocyte; N, neutrophil; Mast, mast cell
TEVs activate stromal cells
Fibroblasts capture TEVs which causes their activation into cancer-associated fibroblasts (CAF). In the liver, TEVs from CRC and hepatocellular carcinoma (HCC) are taken by fibroblasts in metastatic organs and hepatic stellate cells (HSC) in the liver which results in their activation into CAFs through a variety of regulatory mechanisms including miR-controlled protein phosphorylation [55], alteration in lipid metabolism [56] and the production of inflammatory extracellular proteins like Netrin1 [57] and cytokines like IL-6 and IL-8 [58–60]. Studies in lung fibroblasts also demonstrated that TEVs from a large variety of cancers convert fibroblasts into CAFs by activating NFkB and stimulating the production of inflammatory cytokines including IL-6, IL-8 and IL-11 [61–68] and inducing vascular leakiness [69]. TEVs from ovarian carcinoma activate adipose-derived mesenchymal stromal cells (MSC) into CAFs favoring omental metastasis [70, 71]. Fibroblasts capturing TEVs interact with T cells by secreting CCL1 which promotes their differentiation into immunosuppressive Treg [72]. In the brain, astrocytes that can differentiate into fibroblasts, capture melanoma TEVs which activate a proinflammatory signaling and the production of IL-6, IL-8, MCP-1 and CXCL1 promoting brain metastasis [68, 73]. TEVs also stimulate the secretion of IL-1β by astrocytes which induces T cell differentiation into pro-metastatic Th17 cells [74]. The capture of TEVs by epithelial cells is only reported in one publication where it induces the secretion of CCL2 via AKT/NFkB signaling promoting the recruitment of MDSC to the PMN [75].
TEVs stimulate angiogenesis and promote vascular permeability
Microvascular EC are at the frontline in capturing circulating TEVs. Numerous studies in murine and zebrafish models demonstrated that the uptake of TEVs by EC in the microvasculature increases vascular permeability and EC proliferation, creating an abnormal vasculature that facilitates the extravasation of immune cells and tumor cells. TEVs disrupt the formation of tight and adherens junctions between EC by inhibiting the production of cell-cell adhesion molecules such as zona occludens 1, claudin and occludin [76–83] and stimulate endothelial to mesenchymal transition [84]. VE-cadherin is decreased in EC upon capture of TEVs as it is either degraded by a TEV-induced production of the sheddase ADAM-17 [85] or is less expressed [86]. Upon TEV capture, EC initiate ECM remodeling through the production of VEGF, and MMP-2 and MMP-9 [87] and their increased expression of the receptor for urokinase plasminogen activator [88]. TEVs induce in EC the production and secretion of VEGF and chemokines like CX3CL1 (Fractalkine), or CCL2 recruiting immune cells to the PMN [89–91]. In the brain, the capture of TEVs by brain microvascular EC causes a disruption of the blood brain barrier facilitating its crossing by immune and tumor cells [79, 92]. TEV-capturing brain EC affect polarization of microglial macrophages into M2 via Dkk-1 [93]. The mechanisms involved in these profound alterations of the microvasculature are partially known and include a rerouting of the incorporated TEVs from lysosome degradation to endosome processing promoting the expression of angiogenic factors [94] and the activation of signaling pathways such as ERK1/2, AKT and NFkB [79, 89, 95].
TEVs remodel lymph nodes and stimulate lymphangiogenesis
TEVs have a specific organotropism for lymphatic EC. Homing of melanoma exosomes to sentinel lymph nodes results in ECM deposition, lymphatic EC proliferation and melanoma cell recruitment [19]. Through their content in adhesion molecules like α6 integrin [96], CD97 (a G protein coupled receptor) [97] and the nerve growth factor receptor NGFR/p75NTR [98] TEVs promote lymphangiogenesis and lymph node metastasis. The capture of TEVs by lymphatic EC increases their expression of adhesion molecules like ICAM, promoting the homing of circulating tumor cells (CTC) to lymph nodes [98]. Lymphatic EC can act as tumor antigen-presenting cells inducing T cell apoptosis [41]. TEVs are also captured by fibroblastic reticular cells in lymph nodes enhancing PDL-1 expression and T cell exhaustion [99]. TEVs taken by lymphatic EC promote the formation of NETs in the lymph node PMN [100]. The capture of TEVs by macrophages in lymph nodes enhances their secretion of VEGFC stimulating lymphangiogenesis [101].
TEVs remodel the bone
In the bone, TEVs promote an osteolytic PMN and MN through a variety of mechanisms. TEVs and large oncosomes captured by bone marrow macrophages and osteoclasts promote their activation and differentiation in vitro and their osteolytic activity in vivo. They activate in these cells signaling pathways such as NFkB, MAPK, N-WASP and MAFB resulting in an increase in osteolysis [102–107]. TEVs are also captured by osteoblasts stimulating secretory autophagic pathway, increasing inflammation and causing bone remodeling promoting breast and prostate cancer metastasis [108–111]. TEVs are taken by bone marrow MSCs enhancing glycolysis (reverse Warburg effect) and promoting the growth of metastatic cancer cells in the bone marrow [112].
TEVs remodel the ECM
The capture of TEVs by fibroblasts and stromal cells in the PMN and MN remodels the ECM increasing the deposition of ECM proteins like fibronectin, collagen, and tenascin C [113–116]. In an extensive in vivo study of liver metastasis in pancreatic ductal adenocarcinoma (PDAC), investigators demonstrated that TEVs are captured by KC and induce the expression and release of TGFβ which in turn increases the production of fibronectin by HSC in a MIF-dependent mechanism [117]. Other studies have shown that TEVs, in a hypoxic microenvironment, increase the cross linking of ECM proteins via their content in cross linking factors such as lysyl oxidase-like 2 and transglutaminase-2 [115, 118]. ECM remodeling involves not only the synthesis, deposition, and cross-linking of ECM proteins but also their degradation by MMPs secreted by TEV-capturing cells [119, 120].
TEVs enhance the metastatic behavior of tumor cells
TEVs are also captured by tumor cells in a paracrine mechanism. TEVs from high metastatic osteosarcoma cells are captured by low metastatic cells and induce a migratory and invasive phenotype [121] and ADAM-17 in the TEVs of CRC cells cleaves E-cadherin in surrounding tumor cells promoting epithelial to mesenchymal transition and migration [122].
Endogenous models
More recently, innovative labeling strategies and reporter systems that can track TEV release and their capture in mice and zebrafish have been reported [123–126]. Some of these techniques have been combined with endogenous models of TEV release to study their role in the development of the PMN and MN.
In mice bearing genetically modified B16F10 melanoma tumors which produce TEVs carrying membrane-bound Gaussia luciferase, Pucci et al., combined imaging and genetic analysis to track endogenously released TEVs at organismal, cellular, and molecular scales. They show that TEVs efficiently disseminate via lymphatics and preferentially bind to CD109^+^ macrophages in the subcapsular sinus of lymph nodes where they are blocked, indicating that these macrophages have an anti-tumorigenic activity. This blockage however is disrupted upon tumor progression or treatment with chemotherapy (paclitaxel or carboplatin) or immunotherapy (anti-CSFR1). They further demonstrate that in the absence of CD109^+^ macrophages, TEVs interact with B cells fostering a tumor-promoting humoral immunity [18].
Using an elegant Cre-Lox P system in which the capture of TEVs by cells was imaged in vivo by their change in fluorescence (from red to green), Zomer et al. provided evidence for a tumor cell-to-tumor cell TEV transfer process during which TEVs released by malignant cells are taken by less malignant tumor cells located within the same primary tumor and within distant tumors. They show that these TEVs carry mRNAs involved in migration and metastasis [124]. Using PC3 cells stably transduced with a lentivirus containing a fusion of CD63-GFP (PC3-ExGFP) injected into the prostate of male immunodeficient mice, Dai et al. [127] demonstrated the presence of green fluorescence in bone marrow stromal cells three weeks after orthotopic implantation of PC3 cells in the absence of tumor cells, providing the first evidence in an endogenous model in support for the central role of TEVs in triggering the PMN. Using two metastatic models of melanoma and neuroblastoma in immunodeficient mice implanted orthotopically with tumor cells engineered to release GFP-labeled TEVs, our laboratory demonstrated for the first time the capture of TEVs endogenously released in an organotropic manner with a capture in the lung in melanoma and in the liver in neuroblastoma by resident macrophages. As in the report of Dai et al., we observed the presence of TEVs captured by macrophages prior to colonization by tumor cells. The capture of these TEVs by macrophages induced dynamic changes in inflammatory gene expression which evolved to a pro-tumorigenic reaction as the PMN progresses toward the MN [128]. Using an endogenous model of melanoma in immunocompetent mice, Ortiz et al. showed that melanoma TEVs downregulate type I interferon (IFN) receptor and expression of IFN-inducible cholesterol 25-hydroxylase (CH25H). CH25H produces 25-hydroxycholesterol, which inhibits TEV uptake. Mice incapable of downregulating the IFN receptor were resistant to TEV uptake. Treatment of mice with reserpine which decreased the release of TEVs in the plasma of tumor-bearing mice inhibited the formation of lung metastasis. The capture of TEVs in the PMN however was not examined as the TEVs were not labeled [129].
Table 1: Comparison and complementarity between exogenous and endogenous models of TEVs
Conclusion
This review of the literature of the last 15 years demonstrates that exogenous and endogenous models have been complementary in providing experimental evidence that TEVs have an important function in the formation of the PMN and the development of metastasis (Table 1).
Exogenous models have the advantage of their simplicity and the standardization of their methodologies. The high number of TEV-capturing cells that can be harvested and analyzed is another advantage. Exogenous models also have the potential to identify specific subpopulations of TEVs that contribute to metastasis. These models, however, have limitations, particularly due to technical issues related to their in vitro isolation, the methods used for their purification and for their labeling. In particular, the use of lipophilic dyes can result in the formation of aggregates, or micelles, which could mimic EVs and therefore be counted as false-positive signals leading to an overestimation of EV uptake [130–132]. In addition, the doses and schedules used – as demonstrated by our review - widely vary and remain empirical. Endogenous models have the advantage to mimic the natural development of the PMN and its evolution toward the MN as they rely on tumors orthotopically implanted in mice. They are suitable for spatial-temporal analysis of TEVs capture and its effect on the PMN and evolution toward the MN. This advantage has, however, its own limitations due to the implantation of a large number of tumor cells or a tumor fragment. In this regard, mice genetically engineered with a tissue specific fluorescent protein tagged CD63 protein (ExoBow) crossed with genetically engineered mice developing PDAC (KPC model) can establish a true time course development of the PMN from the time of tumor initiation. Authors who developed this model, demonstrated for example the presence of preferential communication routes through exosomes with CAFs and EC in PDAC mice and an increase in inter-organ communication with the kidneys, lungs, and thymus [133]. Endogenous models require engineered cells expressing a chimeric protein made of a fluorescent or luminescent protein and an EV-specific protein, and–based on our experience- the tracking of TEVs and isolation of TEV-capturing cells is more challenging due to their small numbers. Furthermore, these models do not distinguish the different EVs and other non-vesicular components of the secretome involved in the formation of the PMN.
A common limitation to both models is that they have been used to generate gain of function experiments in support of a sufficient role for TEVs in metastasis, leaving the question whether TEVs are necessary for metastasis unanswered. These latter experiments remain challenging as genetic or pharmacological approaches to inhibit or block the production and/or release of EVs and their capture have remained inadequate due to the substantial number of pathways involved in both the biogenesis and the capture of EVs in vivo and their heterogeneity [134, 135]. As our understanding of the mechanisms by which TEVs trigger the PMN and promote the MN continue to increase, it is anticipated that targetable mechanisms will be identified providing an opportunity to design novel approaches to inhibit or prevent metastasis, something that has not been successfully attempted so far and represent an important objective.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3