Structure first – exploration and discovery with cryo-electron microscopy
Miguel Ricardo Leung

TL;DR
Cryo-electron microscopy is transforming structural biology by enabling the discovery of new proteins and interactions at the molecular level.
Contribution
Cryo-EM is enabling a 'structure-first approach' for exploration and discovery of unknown proteins and interactions.
Findings
Cryo-EM allows high-resolution analysis of native protein complexes directly from primary material.
Machine learning and proteomics help identify unknown proteins in cryo-EM maps without prior knowledge.
Cryo-EM is expanding structural biology to uncover new proteins and interactions.
Abstract
The ability to directly observe living systems at finer levels of detail is a strong catalyst for biological discovery. This Perspective highlights how cryo-electron microscopy (cryo-EM) is enabling a ‘structure-first approach’ that can be harnessed for exploration and discovery at the molecular scale, as exemplified in recent studies across the diverse biological contexts curated here. Improvements in throughput, robustness and accessibility of cryo-EM have expanded the range of samples amenable to high-resolution structural analysis to include native protein complexes directly isolated from primary material or imaged unperturbed within the cellular environment. It is therefore increasingly common to encounter unknown proteins in cryo-EM studies, either as unexpected components of a known complex or as completely uncharacterized structures. Advancements in machine learning-assisted…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
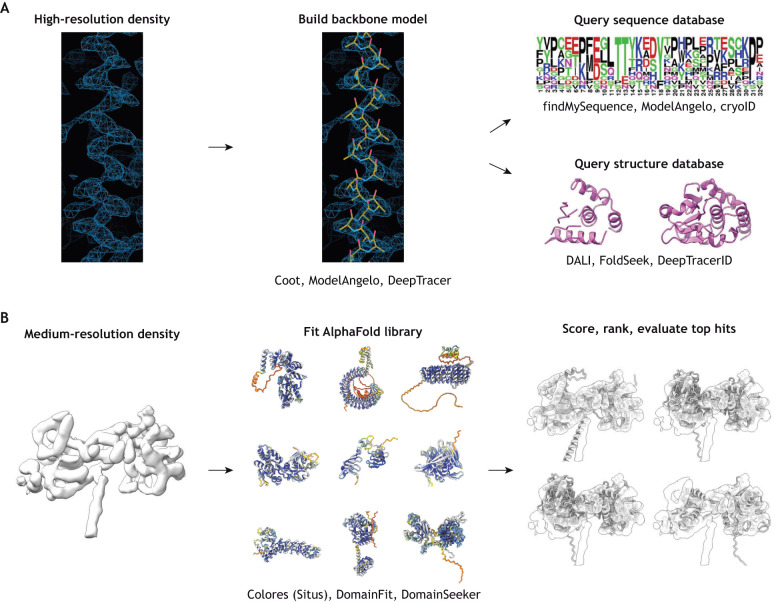 Fig. 1
Fig. 1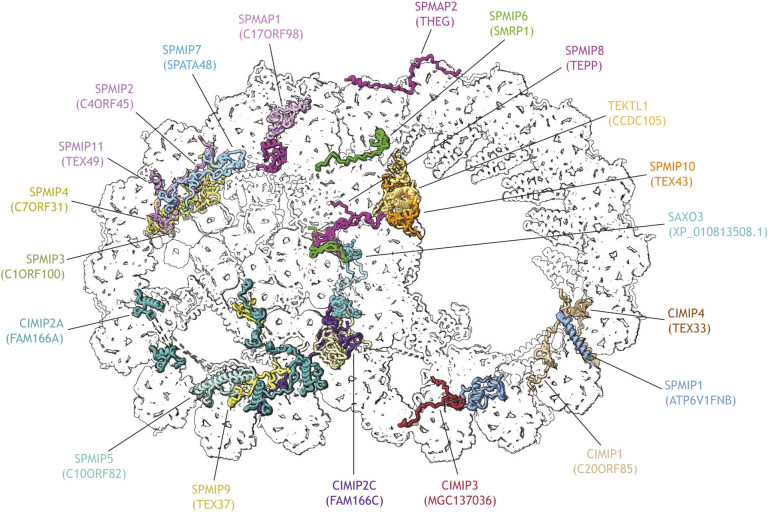 Fig. 2
Fig. 2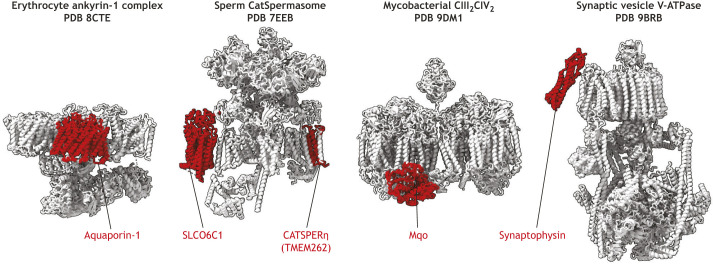 Fig. 3
Fig. 3- —Hubrecht Institutehttps://doi.org/10.13039/501100021800
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Electron Microscopy Techniques and Applications · Electron and X-Ray Spectroscopy Techniques · Enzyme Structure and Function
Introduction
The forward march of biology is driven by the ability to directly observe living systems at ever-finer levels of detail. As Richard Feynman famously stated: “It is very easy to answer many of these fundamental biological questions; you just look at the thing!” (Feynman, 1960). Light microscopes of the 17th century unveiled a world invisible to the naked eye, with pioneering microscopists like Antonie van Leeuwenhoek describing new microscopic organisms nearly everywhere they looked (Wollman et al., 2015). Three centuries later, the electron microscope pulled back the curtain on the complex inner workings of the cell, spurring rapid discoveries of new subcellular structures with astounding variation across different types of cells and tissues (Knott and Genoud, 2013).
In this Perspective, I argue that the electron microscope is an even more powerful discovery tool in the 21st century, supercharged by advancements in cryo-electron microscopy (cryo-EM) and machine learning-enabled protein structure prediction. Cryo-EM enables imaging of biological systems in a near-native state at the molecular scale (Box 1), and incredible progress in the technique over the past ∼10–15 years has expanded the range of samples amenable to high-resolution structural analysis. Perhaps the best-known upside of cryo-EM single-particle analysis (SPA) (Box 1) is that it obviates the need to purify very large quantities of protein and coax them to crystallize. Indeed, cryo-EM is extremely versatile with regards to input material; through image processing and classification, cryo-EM can solve structures of individual components in heterogenous mixtures such as cell lysates, essentially purifying complexes computationally (Arimura et al., 2021; Han et al., 2025; Ho et al., 2020; Kastritis et al., 2017; Kyrilis et al., 2021a; Lyu et al., 2023; Morgan et al., 2022; Sae-Lee et al., 2022; Schmidt et al., 2024; Skalidis et al., 2022; Su et al., 2021, 2023; Tringides et al., 2023; Tüting et al., 2021; Verbeke et al., 2020). More recently, cryo-focused ion beam (cryo-FIB) milling, cryo-electron tomography (cryo-ET) and subtomogram averaging (Box 1) have made it possible to directly visualize proteins within the native cellular environment at molecular or even near-atomic resolutions (Tegunov et al., 2021; Xue et al., 2022). Box 1. Cryo-EM sample preparation and imaging modalitiesCryo-EM (cryo-electron microscopy or electron cryo-microscopy) encompasses a range of techniques that use electrons to image frozen-hydrated biological material at cryogenic temperatures. Samples (proteins, cells or tissues) are frozen so rapidly that ice does not crystallize and instead forms a glass-like or vitreous state. The samples are imaged directly, without heavy metal staining, so signal comes directly from the biological objects. Thus, cryo-EM combines near-native like structural preservation with high-resolution imaging.Whereas proteins, cell lysates and smaller cells can be vitrified directly by rapidly plunging them into liquid cryogen (so-called plunge-freezing) (Dubochet and McDowall, 1981), larger cells and tissues require high-pressure freezing to retard ice crystal nucleation and ensure vitrification throughout the sample volume (Dahl and Staehelin, 1989). Thin samples can be imaged directly by cryo-EM, but samples thicker than ∼1 µm are not suitable for imaging (Baumeister, 2002) because more electrons are lost by inelastic scattering and thus mainly contribute noise to the final image. Larger samples must therefore be thinned by cryo-focused ion beam (cryo-FIB) milling (Marko et al., 2007; Rigort et al., 2012) or by physical sectioning (cryo-electron microscopy of vitreous sections; CEMOVIS) (Al-Amoudi et al., 2004).The main cryo-EM imaging modalities are single-particle analysis (SPA) and cryo-electron tomography (cryo-ET). Both methods aim to generate a faithful three-dimensional (3D) reconstruction from two-dimensional projection images. In SPA, projection images are collected from large numbers of particles, ideally randomly oriented in thin ice (Cheng et al., 2015). The orientations of each particle are determined relative to a reference, and a 3D reconstruction is calculated from the aligned particles. In cryo-ET, projection images are collected from a particular location at different tilt angles, then computationally aligned and back-projected to yield a 3D volume called a tomogram (Turk and Baumeister, 2020). When multiple copies of the same complex are present, subvolumes can be extracted and aligned in a process called subtomogram averaging, enhancing signal-to-noise and increasing resolution (Wan and Briggs, 2016).Traditionally, cryo-EM SPA is applied to purified proteins, whereas cryo-ET and subtomogram averaging are used for more complex systems like cells or organelles. However, this is not a strict distinction – cryo-ET can be performed on (semi-)purified complexes and SPA can also be performed on vesicles and even organelles, cells or cellular lamellae, provided the particle projections can still be reliably aligned despite the presence of overlapping signal. Indeed, many exciting new developments (some described in this article) blur the lines between the techniques or apply them in concert to leverage their individual strengths.
Cryo-EM can therefore yield structural information about targets that were previously intractable. For instance, macromolecular complexes that cannot be recombinantly expressed, reconstituted in vitro or otherwise purified to homogeneity can instead be gently isolated from primary material or imaged directly within intact or partially disrupted cells or organelles. Because protocols with fewer manipulation steps are more likely to preserve fragile protein interactions, a consequence of working with native material is that it becomes increasingly common to encounter unknown proteins, either as unexpected components of a known complex or as completely unidentified structures in raw images or tomograms. Fortunately, it is not necessary to know the identity of a protein complex to reconstruct it in 3D. The main requirements are being able to recognize the complex in raw data, to image many instances of the complex and to align these images computationally. With a cryo-EM map in hand, the task of identifying unknown proteins can be relatively straightforward thanks to an expanding suite of computational approaches, including machine learning-based tools, such as AlphaFold (Jumper et al., 2021).
This leads to a ‘structure-first approach’ in which a cryo-EM map is first calculated, after which molecular composition can be determined directly from the reconstruction. Importantly, this approach requires no prior knowledge of the molecules being reconstructed, and does not necessarily require specific labelling, making cryo-EM of native samples a powerful and versatile approach for protein discovery applicable even to biological systems that have limited molecular or genetic tools available. This Perspective article summarizes general strategies for identifying unknown proteins in cryo-EM maps, then highlights recent examples of how cryo-EM of native samples has led to the discovery of new proteins and interactions in diverse biological contexts.
Strategies for identifying unknown proteins from cryo-EM density maps
The most efficient strategy for identifying proteins de novo from a cryo-EM map depends primarily on what features are visible in the reconstruction (Fig. 1). At low resolutions (>10 Å; 1 Å=0.1 nm), only the overall shape of the protein can be visualized; at intermediate resolution (∼5–7 Å), secondary structure elements can be traced; at high resolutions (below ∼4 Å), amino acid side chains can be resolved, with bulky side chains distinguishable at ∼3–4 Å and smaller side chains distinguishable at ∼3 Å or better.
Workflows for assigning proteins to unknown densities in cryo-EM maps. (A) For high-resolution regions, a backbone model is built into the map. Side chain densities at each position are assessed and used to query a sequence database to find the most likely candidate. Alternatively, the backbone model can be used to directly query a structure database to find proteins with similar folds. (B) For medium-resolution regions, a library of known or predicted structures can be rigid body-fitted into the map. The fits can be scored and ranked or otherwise assessed to determine the best fit. Images in B are published from Leung et al., 2025, where they were published under CC-BY 4.0 terms. Particular packages used at each stage are noted.
When side chain densities can be distinguished, it is possible to use a sequence-based approach (Fig. 1A). Here, the protein backbone is traced either manually, using modelling software like Coot (Emsley et al., 2010), or automatically, using machine learning-based tools like DeepTracer (Pfab et al., 2021), CryoNet (Xu et al., 2019) or ModelAngelo (Jamali et al., 2024). Programs like findMySequence (Chojnowski et al., 2022), cryoID (Ho et al., 2020) or ModelAngelo itself can then be used to assess side chain density and identify the most likely candidate from a sequence database. Alternatively, DeepTracer and ModelAngelo also predict the sequences of modelled fragments, which can be used as input for a separate basic local alignment search tool (BLAST) search. Experienced structural biologists might also be able to infer sequence motifs directly from characteristic shapes of side chain densities (Jiang et al., 2022; Khalifa et al., 2020; Schweighauser et al., 2022), but the aforementioned programs make this process more automated and more objective.
In intermediate-resolution maps where secondary structure elements are visible, fold-based approaches have proven effective means to identify proteins (Fig. 1B). In this strategy, programs like DomainFit (Gao et al., 2024), DomainSeeker (Lu et al., 2024 preprint) or the colores tool in the Situs package (Chacón and Wriggers, 2002; Chen et al., 2023a; Wriggers et al., 1999) are used to automatically rigid body fit (i.e. to directly fit without deformation) a library of protein structures into the unknown density, then to score and rank the top hits. Note that fold-based approaches can also be used for high-resolution maps (Fig. 1A); in these cases, the initial backbone trace can be compared to a database of structures through protein comparison servers, such as DALI (Holm, 2022; Holm and Rosenström, 2010), DeepTracer-ID (Chang et al., 2022b) or FoldSeek (van Kempen et al., 2024). Machine learning-based protein structure prediction tools like AlphaFold make these approaches significantly more powerful by providing extensive libraries of high-quality models against which the unknown density can be queried (Jumper et al., 2021; Varadi et al., 2024). Some studies have also successfully identified unknown proteins from cryo-EM maps using the MOLREP-BALBES pipeline, which uses the program MOLREP to fit domains from the BALBES database containing a curated set of unique folds represented in the Protein Data Bank (PDB) (Brown et al., 2015; Ghanim et al., 2021; Ma et al., 2019), or the Omokage search tool, which compares an unknown structure to PDB or Electron Microscopy Data Bank (EMDB) entries based on overall shape similarity (Skalidis et al., 2022; Suzuki et al., 2016).
Although the above approaches can in principle fish out the correct protein from sequence or structure databases derived from the whole proteome of an organism, it is often very useful to have orthogonal proteomics or cross-linking mass spectrometry data for the specific sample used for cryo-EM (Leung et al., 2025; You et al., 2023). This can greatly facilitate protein identification both by decreasing computational requirements through limiting the subset of candidate proteins and hence the size of the database to search against and can help discriminate between multiple candidate proteins in cases of ambiguity. Approaches synergizing gentle biochemical separation with mass spectrometry of discrete fractions have enabled high-resolution analysis of individual protein assemblies from complex cell lysates (Ho et al., 2020; Kastritis et al., 2017). Proteomics and cross-linking mass spectrometry can also help to identify unknown proteins or to model interactions in maps where resolution is insufficient for unambiguous assignment by either fold-based or sequence-based identification alone, which can be the case with structures derived from in situ cryo-ET due to the overall lower throughput and technical difficulty of the method (O'Reilly et al., 2020; Xing et al., 2024).
Recent examples of structure-guided protein discovery
Microtubule complexes
One of the most bountiful applications of structure-guided protein discovery has been the mapping of highly complex multi-protein networks associated with axonemal microtubules of cilia and flagella in a wide range of species and cell types (Chen et al., 2023a; Doran et al., 2025; Gui et al., 2021, 2022; Ichikawa et al., 2019; Khalifa et al., 2020; Kubo et al., 2023; Leung et al., 2023, 2025; Ma et al., 2019; Stevens et al., 2025; Walton et al., 2023; Xia et al., 2025; Zhao et al., 2025; Zhou et al., 2023; Zhu et al., 2025). Building on pioneering cryo-ET studies in the 2000s that revealed the overall architecture of axonemal microtubules and associated complexes (Nicastro et al., 2006, 2011), these more recent studies produced high-resolution maps that allowed the authors to assign many proteins de novo – in some cases over 100 proteins in a single structure (Leung et al., 2025; Walton et al., 2023; Xia et al., 2025).
In most cases, the authors used cryo-EM single-particle analysis (SPA) of axonemes that been gently disrupted to splay or spread apart the microtubules through mild protease digestion, ATP-induced sliding disintegration or surface tension effects from blotting, although Chen et al. (2023a), Tai et al. (2023), and Zhu et al. (2025) achieved their reconstructions using cryo-ET and subtomogram averaging of cryo-FIB milled sperm flagella. Because axonemal microtubules are large, local resolution varied across the cryo-EM maps and necessitated the use of both sequence- and fold-based approaches to build comprehensive atomic models. Regulatory complexes like the T-shaped radial spokes extending from the microtubule surface were resolved at ∼5–8-Å resolution, likely due to flexibility, so their protein composition was elucidated mainly through fold-based approaches. These strategies were successful in part because regulatory complexes consist mainly of proteins with clear globular domains whose shapes can be resolved even at intermediate resolutions. In contrast, regions closest to the microtubule were better resolved – often at <4 Å – which allowed sequence-based identification of microtubule inner proteins (MIPs), which bind to the microtubule lumen, and microtubule-associated proteins (MAPs), which bind to the external microtubule surface. This is fortunate because most MIPs and MAPs lack clear globular domains and would therefore be difficult to identify unambiguously with fold-based approaches. Indeed, more MIPs and MAPs were able to be identified in reconstructions from SPA, which reached <4 Å, compared to those from subtomogram averaging, which reached resolutions between 4.5 and 6.5 Å.
Together, these structures uncovered the precise binding sites of known proteins implicated in ciliopathies and infertility. More excitingly, they also revealed the identities and interaction networks of previously unknown proteins that represent novel candidate genes associated with male infertility. Among these novel proteins are completely uncharacterized proteins whose uninformative placeholder names (e.g. C#ORF#) reflect the paucity of information about their function or localization. These proteins can now be annotated as microtubule-binding proteins and have thus been renamed ciliary or sperm microtubule inner proteins (CIMIPs or SPMIPs) or microtubule-associated proteins (CIMAPs or SPMAPs), depending on their tissue distribution and their specific localization relative to the microtubule lumen (Leung et al., 2023) (Fig. 2).
Previously uncharacterized proteins identified in the cryo-EM map of the 48-nm repeat of native axonemal doublet microtubules from bovine sperm. The axonemal doublet microtubule structure is from PDB 8OTZ. Uncharacterized proteins with uninformative placeholder names were reclassified as either ciliary microtubule inner proteins (CIMIPs), sperm microtubule inner proteins (SPMIPs), or sperm microtubule associated proteins (SPMAPs).
A particularly striking finding from the structure of mammalian sperm axonemal microtubules was the discovery of an unexpected binding site for an otherwise well-studied complex not previously thought to associate with the axoneme. Cryo-ET of mammalian sperm flagella initially found that the axonemal radial spokes anchored a prominent barrel-shaped complex, which was not found in the axonemes of any other cell type or species studied (Gadadhar et al., 2021; Leung et al., 2021) and was later shown to be distributed asymmetrically around the axoneme (Chen et al., 2023b). Shortly thereafter, cryo-EM SPA of disintegrated bovine sperm flagella resolved the barrel to ∼7.5 Å, which revealed it to be a fully assembled T-complex protein ring complex (TRiC) chaperone complex owing to the distinct shape of the TRiC complex and of its constituent subunits (Leung et al., 2025). This finding raises questions about whether axoneme-tethered TRiC functions in its canonical role as a chaperone or instead plays a mechanoregulatory role by subtly modifying the flagellar beat.
It is difficult to intuit how such a wealth of information could be derived in any other way as efficiently as through cryo-EM-based de novo protein identification. Although several axonemal proteins have been identified in genetically tractable model organisms, such as Chlamydomonas or Tetrahymena, by integrating cryo-ET with gene disruption or tagging approaches (Dymek et al., 2019; Fu et al., 2018, 2019; Gui et al., 2019; Urbanska et al., 2015), these strategies are difficult to apply to mammalian cilia and sperm flagella. For example, mature sperm are transcriptionally and translationally silent, and the only way to perform genetic perturbations in mature sperm is to generate knockout animals. Furthermore, there is no guarantee that disrupting a single protein causes the corresponding loss of a defined axonemal substructure to which the protein can be mapped.
The structures of axonemal microtubules illustrate how cryo-EM can uncover new proteins binding to known scaffolds, in this case to the microtubule lattice. The success of this field is largely thanks to the fact that axonemal complexes bind in various periodicities that are all in coherent register with the 8-nm repeat of tubulin dimers, the fundamental units of the microtubule. Indeed, cryo-EM has also been used to identify novel microtubule-binding proteins in other contexts, such as in cortical microtubules of the parasite Toxoplasma (Wang et al., 2021). To achieve this, the authors treated Toxoplasma cells with detergent and directly imaged the resulting cytoskeletons; microtubules could be successfully picked and aligned despite high background noise from cell debris remaining in the sample.
Filamentous assemblies
Filamentous structures, like microtubules and other cytoskeletal elements, are ubiquitous in biology. Because their striking appearance makes them easy to spot on a cryo-EM grid, it is not entirely uncommon to encounter a filament of unknown composition in a native sample. For example, Cheng et al. (2023, 2024) purified extracellular fibrils from bacterial biofilms using minimal strategies involving only centrifugation and concentration, then solved structures of two unidentified fibrils using cryo-EM SPA. In both cases, the authors used ModelAngelo to automatically build models into high-resolution maps, then identified the proteins through BLAST searches using ModelAngelo-predicted sequences as queries. In a similar vein, Wang et al. (2024a,b, 2025) simply filtered and concentrated pondwater and found diverse filaments with an assortment of shapes and sizes. Without any further fractionation, they used cryo-EM SPA to solve structures of <4 Å for some of these fibrils from an otherwise heterogenous sample. Possibly because of their low abundance and the sheer complexity of the initial sample, these filaments could not be conclusively identified, as no matching sequences were found from proteomics or metagenomics. This challenge highlights an important space for method development that, when addressed, promises to open the door to cryo-EM-guided exploration of environmental samples.
A recent study by Hugener et al. (2024) presented a workflow for identifying unknown filaments that synergizes cryo-ET and subtomogram averaging with cryo-EM SPA. The authors first performed cryo-ET on cryo-FIB-milled lamellae of nutrient-starved yeast cells undergoing gametogenesis and observed uncharacterized filaments in various cellular compartments. They then gently lysed or spread yeast spheroplasts (obtained by treating cells with an enzyme to digest the cell wall) or isolated mitochondria onto cryo-EM grids, which preserved the structure of the filaments while also decreasing sample thickness. This allowed them to collect both cryo-ET and cryo-EM SPA data without the need for tedious FIB milling. The authors used cryo-ET and subtomogram averaging to provide initial low-resolution structures of the filaments that they could then use as initial models to derive helical parameters for subsequent cryo-EM SPA. One type of filament, initially resolved to ∼7 Å, was identified by manually inspecting AlphaFold predictions of mitochondrial proteins that were also upregulated during gametogenesis based on proteomics data; another type of filament, resolved to 3.5 Å, was identified through a DALI search using a manually traced backbone model.
Membrane proteins
Cryo-EM has led to the identification of new components of membrane protein complexes, which have traditionally been very challenging structural targets (Fig. 3). Vallese et al. (2022) purified the native ankyrin-1 complex from erythrocyte membranes and solved its structure to <3 Å, which allowed them to unambiguously identify aquaporin as an unexpected component of the complex. In a creative approach combining genetic manipulation with cryo-EM of native samples, Lin et al. (2021) purified the CatSper channel complex, which mediates the influx of Ca^2+^ necessary for sperm hyperactivation, directly from testes and epididymides of transgenic mice expressing a tagged version of one of the known channel subunits. The authors purified the native CatSper complex through affinity purification and found several unexplained densities, which through mass spectrometry they identified as novel auxiliary subunits. This observation could explain why previous attempts to reconstitute the complex were unsuccessful.
Selected examples of newly identified components of native membrane protein complexes. Aquaporin-1 was identified as a component of the ankyrin-1 complex purified from red blood cells (PDB 8CTE). SLCO6C1 and CATSPERη (formerly TMEM262) were described as novel components of the CatSper channel complex (the ‘CatSpermasome’) from mouse sperm (PDB 7EEB). The malate:quinone oxidoreductase Mqo was found to bind the complex III–complex IV supercomplex from mycobacteria (PDB 9DM1). Synaptophysin was identified as a binding partner of V-ATPase from synaptic vesicles (PDB 9BRB).
Because any form of detergent solubilization risks altering the conformations or interactions of membrane proteins, it would be ideal to solve their structures directly within the bilayer. Recently, several groups have solved structures of membrane proteins natively anchored in cell-derived vesicles and encountered unexplained densities that represented previously unidentified complexes or binding partners that were likely dissociated in previous studies using detergent solubilization. Fu and MacKinnon (2024) prepared native membrane vesicles and observed abundant basket-shaped complexes that they identified as flotillin cages by cryo-EM. Coupland et al. (2024) and Wang et al. (2024a) imaged V-ATPase in native synaptic vesicles using cryo-ET and cryo-EM SPA. Both groups found unexplained densities stoichiometrically associated with the transmembrane regions of V-ATPase, which they identified as synaptophysin by systematically fitting AlphaFold predictions of proteins detected in their preparations. Di Trani et al. (2025) prepared inner membrane vesicles from Mycobacterium smegmatitis and affinity-isolated vesicles containing genetically tagged respiratory complexes. The authors found an extra density binding to the complex III–complex IV supercomplex, which they assigned as malate:quinone oxidoreductase (Mqo) by fitting AlphaFold models of known enzymes related to mitochondrial metabolism.
In the studies described above, the use of small vesicles facilitated high-resolution structure determination by allowing the ice on the cryo-EM grids to remain fairly thin, so that high-quality projection images could be collected and analysed by cryo-EM SPA. Another potentially interesting technique for exploring membrane-associated processes is unroofing, in which mechanical shear is applied to cells through, for example, a pressurized fluid. This procedure also washes away most of the cytoplasm, yielding patches of native plasma membrane and associated material that are thin enough to image without cryo-FIB milling (Sun et al., 2025). It even appears to be possible to solve high-resolution structures of membrane proteins from whole organelles by SPA, at least for large complexes such as mitochondria respiratory complexes (Zheng et al., 2024).
There are also encouraging results demonstrating the potential of structure-guided discovery directly in intact cells. In a recent preprint, Jensen et al. (2025) preprint visualized large dome-shaped structures on the plasma membrane of Mycoplasma pneumoniae by cellular cryo-ET. They reconstructed the complex at ∼9-Å resolution by subtomogram averaging and, by fitting AlphaFold models of candidates from surface-shaving proteomics (in which intact cells are gently treated with protease to enrich for surface-exposed proteins) and cross-linking mass spectrometry, identified it as a complex of previously uncharacterized proteins that associate with the protein translocation machinery. As a testament to the discovery power of in situ cryo-ET, they also found that ribosomes associated with the dome complex, hinting that the complex might function as a chaperone-like folding chamber for newly translated and translocated proteins.
Pathogenic elements
An intriguing application of the structure-first approach is the identification of disease-related or disease-causing agents in a form of ‘molecular pathology’. For instance, cryo-EM has been used to identify amyloid fibrils isolated from postmortem human brain as being composed of the unexpected protein TMEM106B (Chang et al., 2022a; Jiang et al., 2022; Schweighauser et al., 2022). Because the cryo-EM maps were resolved at <3 Å, protein identity could be deduced by sequence. Interestingly, the three groups used slightly different approaches but converged on the same solution: Schweighauser et al. used sequence motifs deduced from the map to scan the human proteome; Jiang et al. manually inferred the best-fitting amino acid at each position and used the resulting sequence in a BLAST search; and Chang et al. used automated approaches implementing findMySequence and cryoID.
Cryo-EM was also recently used to identify a virus infecting farmed superworm (Zophobas morio) larvae (Penzes et al., 2024). The authors isolated viruses directly from carcasses of infected larvae and solved cryo-EM structures to <3 Å. Two independent approaches were used to identify the virus. First, models were built either manually or automatically with ModelAngelo and used as inputs to the DALI server to query the PDB. Second, ModelAngelo-predicted sequences were used to query UniProt. Both approaches yielded proteins from the same viral subfamily as top hits, which informed the choice of an appropriate strategy to sequence the whole viral genome. Impressively, information about viral identity was derived from the cryo-EM map within 1 week of receiving the first samples, illustrating that cryo-EM can provide rapid identification times for ideal samples like abundant, highly symmetric viruses.
Challenges and prospects
Cryo-EM and cryo-ET are clearly powerful tools, but there are some important considerations that limit the applicability of the structure-first approach. These considerations are inherent to averaging-based approaches: namely, that the target needs to be at least partially structured or repetitive (i.e. ‘averageable’), and the target needs to be abundant in the sample, because large datasets (typically hundreds of thousands to millions of initial particle images) are often needed to reach relatively high resolutions required for protein identification (<∼10 Å), with better resolutions permitting faster and more confident assignments. In practice, this approach is also currently best-suited to protein complexes that are sufficiently large for their particle images to be recognized and aligned despite the potentially high background noise in partially purified or in situ samples. Fortunately, it is precisely these large supramolecular assemblies that benefit the most from such an approach as they have complex multi-protein compositions that might not be fully elucidated and are otherwise challenging to study given that they can be difficult to reconstitute or purify to homogeneity without losing components. In the near future, these hurdles will become easier to overcome thanks to advances like improved data collection throughput (Cheng et al., 2018; Eisenstein et al., 2023), better microscope hardware (Nakane et al., 2020; Yip et al., 2020) and image processing pipelines (Kimanius et al., 2024; Tegunov et al., 2021), easier access to high-end instrumentation through shared facilities and national centres (Saibil et al., 2015; Zimanyi et al., 2022), and availability of lower-cost 100-kV electron microscopes (Karia et al., 2025; McMullan et al., 2023).
Many of the examples above are from protein complexes that have been at least partially liberated from the cellular context. However, there are examples from completely intact cellular specimens (Chen et al., 2023a; Jensen et al., 2025) and there are certain to be more such examples enabled by maturation of the technologies and workflows for cryo-FIB milling, cryo-ET and subtomogram averaging. Hybrid workflows using cryo-EM SPA on FIB-milled lamellae also show great promise, with the potential to increase data collection throughput on cellular samples (You et al., 2023; Zheng et al., 2025).
Most structure-based protein discovery projects will likely involve integrating in situ cryo-ET and subtomogram averaging with cryo-EM SPA. The initial observation of an unidentified molecular species in cellular cryo-ET data can spur follow-up efforts aimed at reducing the complexity of the system to make targets amenable to cryo-EM SPA while retaining their structures as close as possible to the native state. For abundant targets that are easy to recognize for imaging, gentle disruption through mechanical lysis, hypotonic rupture or mild detergent treatment might suffice. For smaller or rarer targets, it might be necessary to include an enrichment step involving concentration, differential centrifugation, sucrose gradient fractionation or size exclusion chromatography. Once data is collected, even low-resolution maps derived from in situ cryo-ET can be used as initial references for alignment. The resulting high-resolution ex situ maps can also be compared to these low-resolution cellular reconstructions to determine whether any proteins have been lost during purification.
‘Visual proteomics’ strategies, where fractionated cell lysates are analysed by cryo-EM and mass spectrometry, also represent powerful approaches for systematically exploring the molecular composition of cells (reviewed in Klykov et al., 2022; Kyrilis et al., 2019, 2021b; McCafferty et al., 2020; Ziegler et al., 2021). Excitingly, applying this approach to the Tetrahymena ciliary matrix has recently led to the identification and structural characterization of a novel type of protein assembly called the ‘CAGE complex’ that appears to be conserved across broad swaths of the tree of life, encouraging future studies into the functional role of the complex (McCafferty et al., 2025). These studies can likewise complement in situ cryo-ET by providing reference data to interpret highly complex cellular tomograms.
It is important to note that cellular cryo-ET and subtomogram averaging are currently the only methods that can report on the true structures of protein complexes in situ, and on the subcellular distribution of protein complexes at molecular resolution. For example, recent in situ cryo-ET studies have shown that the CatSper channel forms extensive zigzag arrays on the plasma membrane of the mammalian sperm flagellum (Zhao et al., 2022) and that TRiC chaperonin particles in the closed conformation form linear arrays in the cytoplasm (Xing et al., 2024). This information is lost by even the gentlest isolation and purification procedures, but precise knowledge about the spatial arrangement of molecular machines and their conformational states is, in and of itself, an important driver for generating new biological models and hypotheses.
Concluding remarks
The ability of cryo-EM to produce high-resolution structures from native material is leading to the discovery of novel proteins and interactions in diverse systems. This represents an exciting shift in how structural biology contributes to our broader understanding of biological mechanisms – rather than being the final piece of the puzzle that explains prior biochemical data, a cryo-EM map is now often a starting point for hypothesis generation. Because cryo-EM maps simultaneously contain information about protein identity, structure and interactions, they are important resources for rationally designing downstream functional or genetic studies. Thus, cryo-EM-guided protein discovery could prove to be one of the more prominent roles for experimental structural biology in the post-AlphaFold era. Democratizing access to cryo-EM instrumentation and expertise to accommodate a wider range of projects will broaden the application of this structure-first approach from serendipitous discovery to structured exploration.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Al-Amoudi, A., Chang, J.-J., Leforestier, A., Mc Dowall, A., Salamin, L. M., Norlén, L. P. O., Richter, K., Blanc, N. S., Studer, D. and Dubochet, J. (2004). Cryo-electron microscopy of vitreous sections. EMBO J. 23, 3583-3588. 10.1038/sj.emboj.760036615318169 PMC 517607 · doi ↗ · pubmed ↗
- 2Arimura, Y., Shih, R. M., Froom, R. and Funabiki, H. (2021). Structural features of nucleosomes in interphase and metaphase chromosomes. Mol. Cell 81, 4377-4397.e 12. 10.1016/j.molcel.2021.08.01034478647 PMC 8571072 · doi ↗ · pubmed ↗
- 3Baumeister, W. (2002). Electron tomography: Towards visualizing the molecular organization of the cytoplasm. Curr. Opin. Struct. Biol. 12, 679-684. 10.1016/S 0959-440X(02)00378-012464323 · doi ↗ · pubmed ↗
- 4Brown, A., Long, F., Nicholls, R. A., Toots, J., Emsley, P. and Murshudov, G. (2015). Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. D Biol. Crystallogr. 71, 136-153. 10.1107/S 139900471402168325615868 PMC 4304694 · doi ↗ · pubmed ↗
- 5Chacón, P. and Wriggers, W. (2002). Multi-resolution contour-based fitting of macromolecular structures. J. Mol. Biol. 317, 375-384. 10.1006/jmbi.2002.543811922671 · doi ↗ · pubmed ↗
- 6Chang, A., Xiang, X., Wang, J., Lee, C., Arakhamia, T., Simjanoska, M., Wang, C., Carlomagno, Y., Zhang, G., Dhingra, S. et al. (2022 a). Homotypic fibrillization of TMEM 106B across diverse neurodegenerative diseases. Cell 185, 1346-1355.e 15. 10.1016/j.cell.2022.02.02635247328 PMC 9018563 · doi ↗ · pubmed ↗
- 7Chang, L., Wang, F., Connolly, K., Meng, H., Su, Z., Cvirkaite-Krupovic, V., Krupovic, M., Egelman, E. H. and Si, D. (2022 b). Deep Tracer-ID: De novo protein identification from cryo-EM maps. Biophys. J. 121, 2840-2848. 10.1016/j.bpj.2022.06.02535769006 PMC 9388381 · doi ↗ · pubmed ↗
- 8Chen, Z., Shiozaki, M., Haas, K. M., Skinner, W. M., Zhao, S., Guo, C., Polacco, B. J., Yu, Z., Krogan, N. J., Lishko, P. V. et al. (2023 a). De novo protein identification in mammalian sperm using in situ cryoelectron tomography and Alpha Fold 2 docking. Cell 186, 5041-5053.e 19. 10.1016/j.cell.2023.09.01737865089 PMC 10842264 · doi ↗ · pubmed ↗