Diabetes Mellitus Accelerates Alzheimer′s Disease Development by Affecting the Gut Microbiome
Qiong He, Zixiao Zhao, Donglang Jiang, Aihua Fei

TL;DR
This study shows that diabetes worsens Alzheimer's by disrupting gut bacteria, and suggests that butyrate can help reduce brain amyloid buildup.
Contribution
The study reveals a novel mechanism linking diabetes and Alzheimer's through gut microbiome changes and demonstrates butyrate's therapeutic potential.
Findings
Diabetes increases amyloid beta deposition in Alzheimer's mouse models.
Butyrate reduces brain amyloid and improves gut microbiome health.
Gut dysbiosis in diabetic mice is marked by reduced short-chain fatty acid producers.
Abstract
Increasing evidence suggests a link between Alzheimer′s disease (AD) and diabetes mellitus (DM). However, the precise mechanisms by which DM contributes to AD remain unclear. This study is aimed at elucidating the potential role of DM in the early stages of AD. Accordingly, a streptozotocin (STZ)‐induced diabetic 5 × familial AD (FAD) mouse model was established. Immunohistochemistry and positron emission tomography/computed tomography (PET/CT) scanning were performed to examine amyloid beta (Aβ) deposition in the brain. The integrity of the colonic epithelium was assessed using quantitative reverse transcription–polymerase chain reaction (qRT‐PCR) and immunofluorescence staining. Microbial diversity analysis was conducted for 5 × FAD mice with and without STZ‐induced DM to determine shifts in intestinal flora profiles. After oral administration of butyrate to STZ‐treated 5 × FAD mice,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
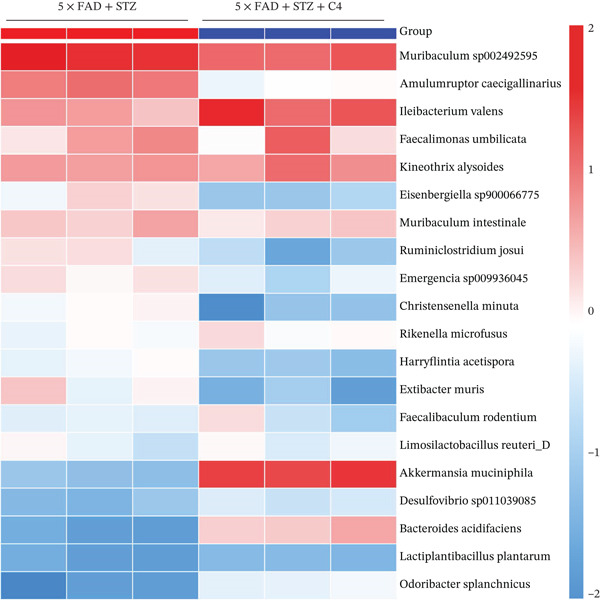 Figure 1
Figure 1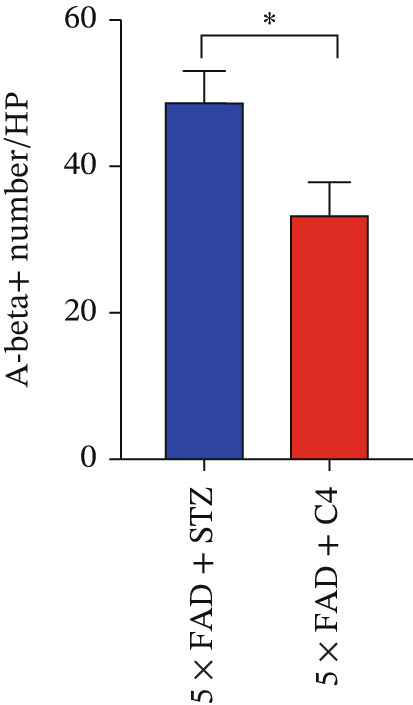 Figure 2
Figure 2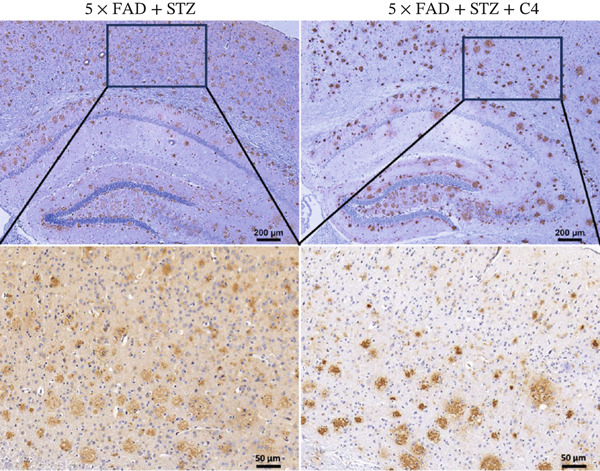 Figure 3
Figure 3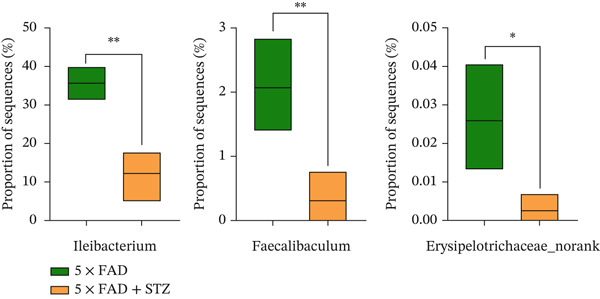 Figure 4
Figure 4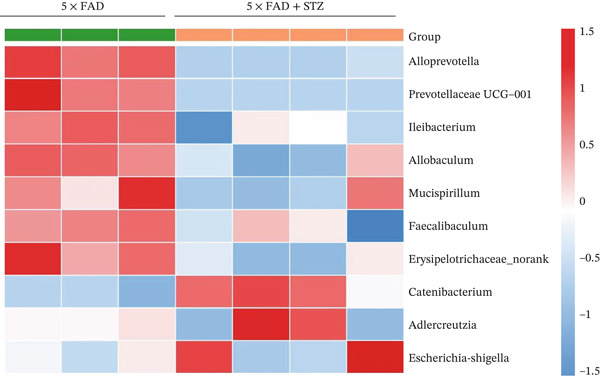 Figure 5
Figure 5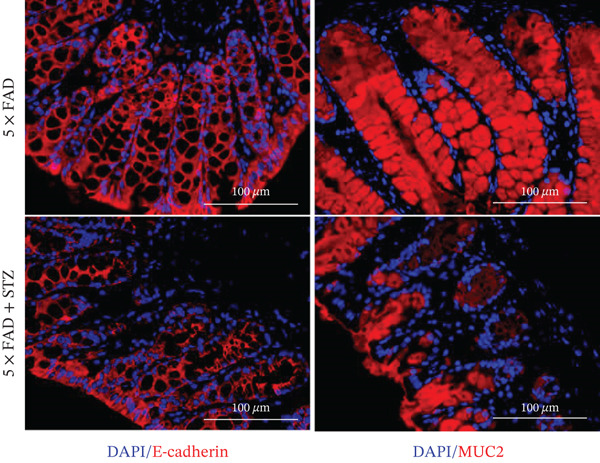 Figure 6
Figure 6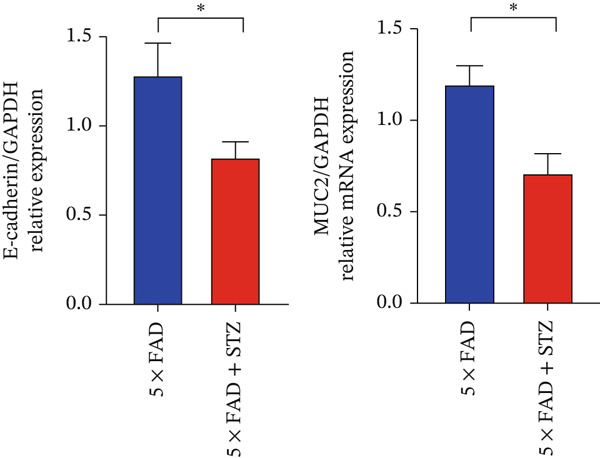 Figure 7
Figure 7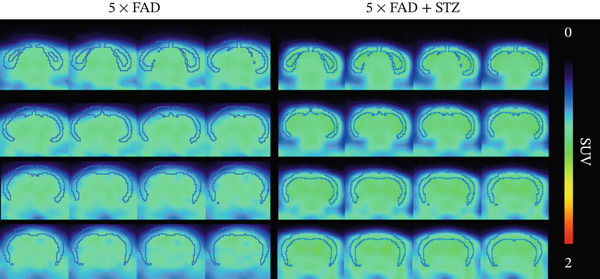 Figure 8
Figure 8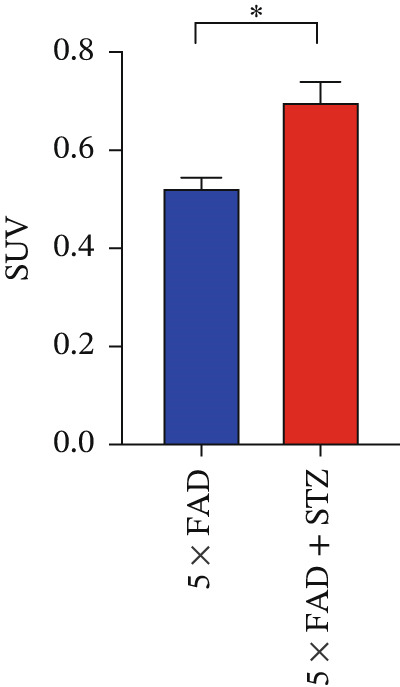 Figure 9
Figure 9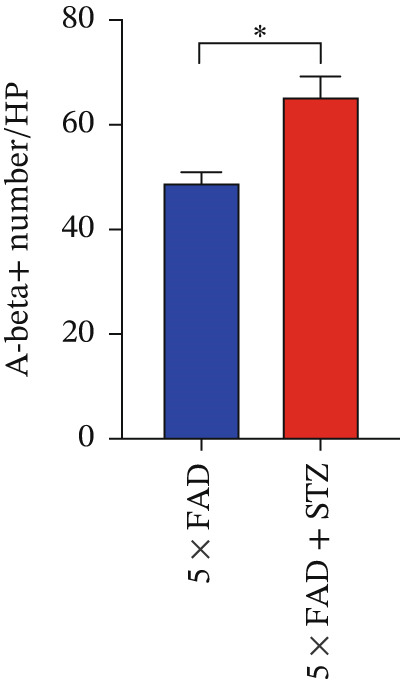 Figure 10
Figure 10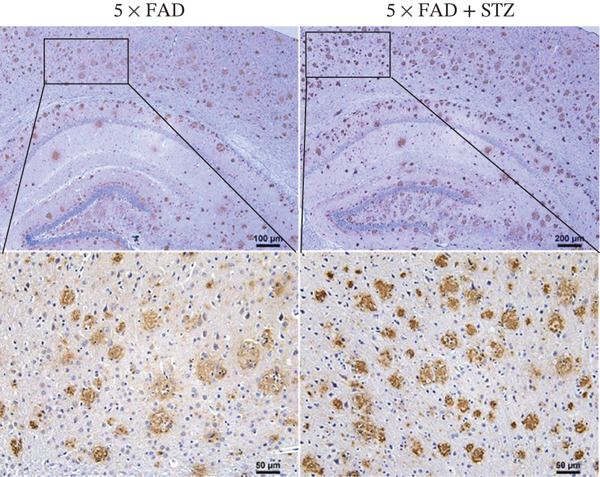 Figure 11
Figure 11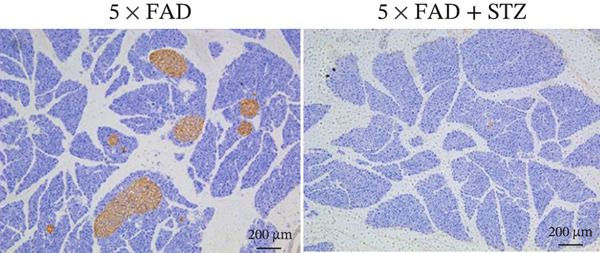 Figure 12
Figure 12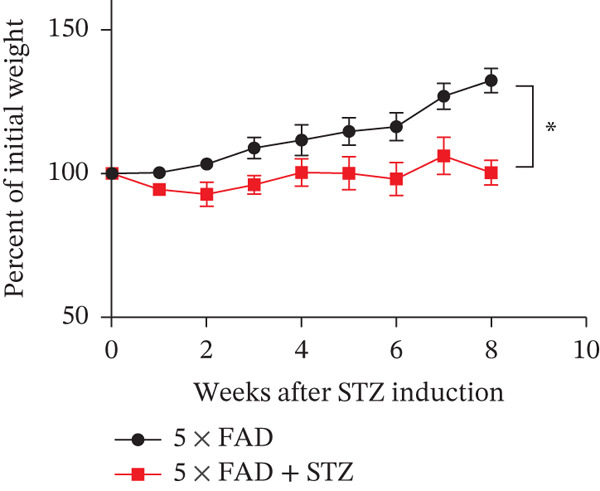 Figure 13
Figure 13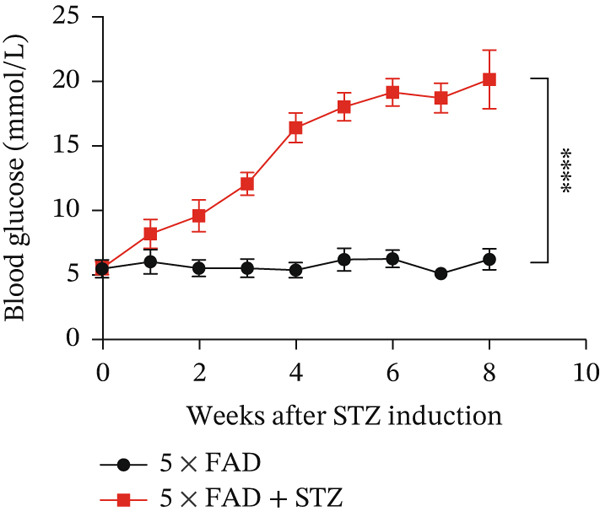 Figure 14
Figure 14- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Immune responses and vaccinations · Alzheimer's disease research and treatments
1. Introduction
Alzheimer′s disease (AD) is a neurodegenerative disorder and the primary cause of dementia in older individuals [1]. In recent years, the prevalence of AD has increased rapidly worldwide. The World Health Organization estimates that approximately 40 million people globally suffer from AD, with this number expected to increase to 78 million by 2030 and 139 million by 2050 [2]. AD is a complex, multifactorial disease. Two main theories have emerged regarding AD pathogenesis: the amyloid hypothesis, which posits that AD is caused by the accumulation of amyloid beta (Aβ) protein, and the cholinergic hypothesis, which suggests that impaired cholinergic function is a critical risk factor for AD [3]. However, the etiology and pathology of AD remain poorly understood. Given the absence of effective treatments and considerable economic and psychological burdens, therapeutics for AD are needed.
Several factors, including genetic mutations, advanced age, obesity, hypertension, and diabetes mellitus (DM), contribute to AD pathogenesis [4]. Genetic studies have indicated that 60%–80% of AD risk is attributable to heritable factors; however, the common apolipoprotein E ε4 allele does not fully account for AD heritability [5]. A large genome‐wide meta‐analysis involving approximately 150,000 individuals with AD, 300,000 individuals with a proxy phenotype of AD, and matched controls identified over 40 novel genetic variants associated with AD, including Phospholipase C Eta 1 (PLCH1), death effector domain containing (DEDD), and WD repeat domain 12 (WDR12) [6]. Aging is a key risk factor for AD, as most cases occur after 65 years of age [7]. Studies on animals and patients with AD have shown that exposure to air pollution can damage the olfactory mucosa and bulb, as well as the frontal cortex, which may contribute to the occurrence of AD [8]. Other factors, such as diet, infections, and medical conditions, are also associated with an increased risk of AD [9].
Metabolic disorders, particularly DM, are closely linked to AD progression [10]. DM is a chronic condition characterized by the inability of pancreatic beta cells to sufficiently secrete insulin and the body′s inability to effectively use insulin, resulting in elevated blood glucose levels. Both DM status and longer DM duration are significantly associated with worse cognitive outcomes, and patients with DM have a two‐ to fivefold increased risk of developing AD compared with the general population [11]. Statistical analyses of patients with mild cognitive impairment and AD have suggested that DM may indirectly contribute to cognitive deterioration through its association with reduced baseline cortical thickness, potentially due to a reduction in brain reserve [12]. However, the biological basis and molecular mechanisms underlying the correlation between these two chronic diseases remain unclear.
The close association between AD and DM may be related to factors such as impaired insulin signaling, inflammatory responses, and oxidative stress, which promote metabolic, vascular, and neurological dysfunction [13]. Insulin‐Like Growth Factor I (IGF‐I) and Insulin‐Like Growth Factor II (IGF‐II) are widely expressed in neurons and glia, including in structures associated with neurodegeneration and those that mediate neurocyte growth, metabolism, survival, and neurotransmitter networks [14]. More precise data have shown that insulin deficiency triggers Aβ plaque accumulation, increased tau phosphorylation, neurofibrillary tangle formation, and memory impairment [15]. Furthermore, the hyperglycemic status in DM mediates specific effects in AD pathogenesis. Hyperglycemia, which occurs in patients with DM, affects K_ATP_ channels, altering neuronal metabolism and increasing Aβ levels in the brain [16]. Clinical studies have confirmed that advanced glycation end‐products, which promote Aβ plaque and neurofibrillary tangle formation, are more abundant in patients with AD and DM than in those with AD alone [17]. Additional crosstalk between AD and DM has also been identified. The mammalian target of rapamycin (mTOR) pathway, a key regulator of neurogenesis that enhances the expression of brain‐derived neurotrophic factor, is hyperactivated in both AD and DM, exacerbating cognitive decline. Aβ injection impairs synaptic plasticity associated with mTOR signaling [18], which may involve the PI3K/Akt/mTOR signaling pathway. However, given the unsatisfactory outcomes of treatment for AD, further research is needed to unravel the molecular mechanisms underlying the relationship between AD and DM. Therefore, in the present study, we aimed to develop a mouse model of AD with inducible DM to assess the role of DM in AD pathogenesis in the brain and other organs.
2. Materials and Methods
2.1. Mice
We employed the 5 × FAD mouse model, a widely used model for AD research [19]. Male 5 × FAD mice were purchased, and the genotypes were confirmed by the Shanghai Model Organisms Center, Inc. (Shanghai, China). All mice were bred and maintained under specific pathogen‐free conditions at the Experimental Animal Centre of Tongji University School of Medicine (Shanghai, China). All mice were allowed free access to filtered air, sterile water without antibiotics, and autoclaved food in individually ventilated cages with a 12‐h light‐dark cycle. Animals aged 8–10 weeks were used for the experiments. All animal research protocols were approved by the Animal Welfare and Ethics Committee. This study was carried out in compliance with the ARRIVE guidelines.
2.2. Induction of DM Using STZ
DM was induced in 8–10‐week‐old male 5 × FAD mice using high‐dose STZ, as described previously [20]. Briefly, STZ (Sigma‐Aldrich, St. Louis, Missouri, United States) was injected intraperitoneally (150 mg/kg, 0.1 M citrate buffer) on the first day, whereas the control group was injected with citrate buffer (7.5 mL/kg, Sigma‐Aldrich). DM was confirmed in the fourth week by a high glucose level of ≥ 11.1 mmol/L after a 12‐h fast. Body weight was monitored weekly. All mice were sacrificed at the end of the experiments.
2.3. Administration of C4
STZ‐5 × FAD mice were categorized into treatment and control groups. According to previous studies [21], the treatment group was given C4 (200 mM, Sigma‐Aldrich) in drinking water from Day 0 until the end of the experiment. The control group was given sterile water orally.
2.4. Immunohistochemical Staining
Mice were anaesthetized, and cardiac perfusion of 4% paraformaldehyde was performed. The brains and pancreas were removed and sectioned into 5‐μm slices. Following deparaffinization and rehydration, the slides were incubated with 0.3% Triton X‐100 for 10 min and blocked with 10% normal rabbit serum for 1 h at 25°C. Immunohistochemistry was performed using either mouse anti‐insulin monoclonal antibody (mAb) (Servicebio, Wuhan, China; 1:500) or rabbit antibeta amyloid mAb (Abcam, Cambridge, United Kingdom; 1:1000) overnight at 4°C. Negative controls were prepared using the same procedures, with normal serum replacing the antibodies. The following day, the slides were washed and incubated with goat antimouse immunoglobulin G (IgG) (Servicebio; 1:400) or donkey antirabbit IgG (Servicebio; 1:400). All slides were assessed using a 3,3 ^′^‐diaminobenzidine Substrate Kit (Servicebio).
2.5. Immunofluorescence Staining
Samples were fixed with 10% paraformaldehyde, embedded in paraffin, and sectioned into 5‐μm slices. Following deparaffinization and rehydration, slides were incubated with 0.3% Triton X‐100 for 10 min and blocked with 10% normal donkey serum for 1 h at room temperature. Immunostaining was performed using mouse anti‐E‐cadherin mAb (Servicebio; 1:500)/rabbit anti‐mucin 2 (MUC2) mAb (Servicebio; 1:500) overnight at 4°C. Negative controls were prepared using the same procedures, with normal serum replacing the antibodies. The following day, the slides were incubated with goat antimouse IgG‐CY3 (Servicebio; 1:300) or goat antirabbit IgG‐CY3 (Servicebio; 1:300) for 1 h at room temperature. Coverslips were subsequently mounted onto glass slides using a Slowfade Gold antifade mountant, and 4 ^′^,6‐diamidino‐2‐phenylindole (Servicebio; 1:1000) was used to counterstain the DNA. The images were captured using a fluorescence microscope.
2.6. Micro–Positron Emission Tomography/Computed Tomography (Micro‐PET/CT)
Micro‐PET/CT was performed for the mice as described previously [22]. Briefly, the mice were anaesthetized with 2% isoflurane in 100% oxygen (1 L/min) at room temperature using an isoflurane vaporizer (Molecular Imaging Products Company, United States). The mice were placed in a spread‐supine position on the imaging bed and administered 1% isoflurane in 100% oxygen (1 L/min) during PET/CT. Static [^18^F]‐AV45 PET imaging was conducted for 10 min at 30 min after the administration of [^18^F]‐florbetapir (AV45; ∼5 μCi/g body weight). PET images were reconstructed using the ordered subset expectation maximization three‐dimensional algorithm, and the data were reviewed and processed using IRW. Image processing was performed using the PMOD software (Version 4.4, PMOD Technologies Ltd., Zurich, Switzerland). Images were manually fused with the PMOD mouse template, and data from 19 regions of interest in the brain were collected. The mean standardized uptake value of the cortex was calculated.
2.7. Quantitative Reverse Transcription–PCR (qRT‐PCR)
Total RNA was extracted using the TRIzol reagent (Life Technologies, Grand Island, New York, United States). Complementary DNA was subsequently synthesized using a 5× All‐In‐One RT MasterMix Kit (Applied Biological Materials, Richmond, Canada) according to the manufacturer′s instructions. PCR amplification was performed in triplicate under the following conditions: 25°C for 1 min, 42°C for 15 min, and 85°C for 5 min. A 7900HT Fast Real‐Time PCR system (Applied Biosystems, Carlsbad, California, United States) and TB Green Premix Ex Taq PCR Kit (TaKaRa, Dalian, China) were used for qRT‐PCR, with cycling conditions of 95°C for 30 s and 40 cycles of 95°C for 5 s and 60°C for 30 s (primers are shown in Table S1). The relative expression levels of the target gene were normalized to the housekeeping gene GAPDH and calculated using the 2^−△△Ct^ method.
2.8. Microbial Diversity Analysis
The microbial diversity in mouse fecal samples was evaluated using 16S rRNA gene sequencing. Microbial DNA was isolated using the E.Z.N.A. Soil DNA Kit (Omega Bio‐tek, Norcross, Georgia, United States). The bacterial 16S ribosomal RNA genes were amplified through PCR using the following cycling conditions: denaturation at 98°C for 2 min, 27 cycles of 30 s of denaturation at 95°C, 30 s of annealing at 55°C, and 60 s of extension at 72°C; a final extension at 72°C for 5 min was performed. Each PCR mixture was 20 μL, containing 4 μL of 5× FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. SMRTbell libraries were prepared using blunt‐end ligation following the manufacturer′s protocol (Pacific Biosciences, San Diego, California, United States), purified from Zymo and HMP mock communities, and sequenced using PacBio Sequel II 8 M cells with Sequencing Kit 2.0 (Shanghai Biozeron Biotechnology Co., Ltd.). The circular consensus sequences obtained were filtered and clustered into operational taxonomic units with a 98.65% similarity threshold using UPARSE (Version 7.1), ensuring at least three passes and a predicted accuracy of 0.99. Biomarkers between groups were identified using the linear discriminant analysis effect size. Significance was set at p < 0.05, with linear discriminant analysis scores ≥ 2.0 for the Kruskal–Wallis and pairwise Wilcoxon tests. p values were adjusted using the false discovery rate.
2.9. Statistical Analysis
Data were processed using GraphPad Prism 8 (GraphPad Software Inc., San Diego, California, United States). To assess significance, unpaired or paired Student′s t‐test and one‐way analysis of variance were employed. Results are presented as the mean ± standard error of the mean, with the threshold for significance set at p < 0.05.
3. Results
3.1. High Glucose Aggravates Aβ Deposition in the Brains of 5 × FAD Mice With Induced DM
The successful establishment of the STZ‐induced DM model in 5 × FAD mice was verified by elevated blood glucose levels (≥ 11.1 mmol/L) (Figure 1a). Notably, 5 × FAD mice treated with STZ showed a significant loss in body weight (Figure 1b). Simultaneously, insulin staining of pancreatic tissue was performed, which confirmed the lack of insulin expression in the DM group (Figure 1c).
Figure 1. High glucose aggravates Aβ deposition in the brain. n = 4 for each group. (a) The blood glucose levels of 5 × FAD mice that received STZ or citrate buffer were monitored weekly. ^∗∗∗∗^ p < 0.0001 vs. 5 × FAD mice that received citrate buffer. (b) The body weight of 5 × FAD mice that received STZ or citrate buffer was monitored weekly. Changes in body weight were indicated as a percentage of the original weight at the start of the experiments. ^∗∗^ p < 0.01 vs. 5 × FAD mice that received citrate buffer. (c) Immunohistochemical staining for insulin of representative sections from pancreatic tissue of 5 × FAD mice that received STZ or citrate buffer (original magnification ×50). (d) Immunohistochemical staining for Aβ of representative sections from brain tissues of 5 × FAD mice that received STZ or citrate buffer (upper: original magnification ×50; lower: original magnification ×200). (e) Bar chart of the number of Aβ ^+^ cells in each HP view, ^∗∗^ p < 0.01 vs. 5 × FAD mice that received citrate buffer. (f) Statistical analysis and comparison of the cortical SUV in 5 × FAD and 5 × FAD mice with STZ treatment ( ^∗^ p < 0.05). (g) Representative [^18^F]‐AV45 uptake according to micro‐PET/CT imaging in 5 × FAD with citrate buffer and 5 × FAD mice with STZ treatment. Brain [^18^F]‐AV45 uptake was higher in the cortex of 5 × FAD mice with STZ treatment than in 5 × FAD mice with citrate buffer treatment. Regions of interest drawn in the cortex (blue) are displayed on each image.(a)(b)(c)(d)(e)(f)(g)
Given the strong correlation between DM and AD, we investigated the potential role of DM in AD progression. Brain tissues from 5 × FAD mice with STZ treatment showed increased numbers of Aβ plaques compared with those without STZ treatment (Figure 1d,e). As [^18^F]‐AV45 is a PET radiopharmaceutical agent pathologically confirmed to detect Aβ plaques in brain tissue [23], we performed PET brain imaging with [^18^F]‐AV45 for all mice. Compared with control mice, 5 × FAD mice with STZ showed significantly increased [^18^F]‐AV45 uptake in the cortex (0.701 ± 0.090 vs. 0.525 ± 0.050, p = 0.029), indicating increased amyloid deposition (Figure 1f,g).
3.2. DM Worsens Intestinal Barrier Dysfunction in AD Mice and Induces Changes in the Intestinal Flora of AD Mice
The integrity of the colonic epithelium was investigated by examining the mRNA and protein expressions of MUC2 and E‐cadherin in colon tissue using qRT‐PCR and immunofluorescence staining. The results confirmed decreased MUC2 and E‐cadherin expression in 5 × FAD mice treated with STZ than in the controls (Figure 2a,b), suggesting that DM exacerbates intestinal barrier dysfunction in AD.
Figure 2. High glucose drives gut barrier damage in 5 × FAD mice and disrupts the gut microbial homeostasis. (a) Colon tissues were obtained, and RNA was extracted. ^∗^ p < 0.05. n = 4 for each group. The data shown are representative of three independent experiments with four mice per group. (b) Representative immunofluorescent images of colonic mucosa for staining with DAPI (blue) and anti‐E‐cadherin (red [left]) or anti‐MUC2 (red [right]). Original magnification ×400. (c) The feces of two groups were collected, and 16S ribosomal DNA sequencing was performed. n = 3 for 5 × FAD mice that without STZ treatment and n = 4 for 5 × FAD mice that received STZ treatment. A heatmap of the top 10 different microbiota species between the two groups was exhibited. (d) Relative abundance of Ileibacterium, Faecalibaculum, and Erysipelotrichaceae_norank in two groups. Comparisons between multiple groups were analyzed using Student′s t‐test. ^∗^ p < 0.05 and ^∗∗^ p < 0.01.(a)(b)(c)(d)
We then examined the fecal microbiomes of the 5 × FAD mice. These mice exhibited more severe gut dysbiosis than the controls, characterized by increased harmful bacteria such as Escherichia–Shigella and reduced beneficial bacteria such as Ileibacterium and Faecalibaculum (Figure 2c). This dysbiosis was also reflected in changes in the proportion of bacteria involved in short‐chain fatty acid (SCFA) synthesis and metabolism. Significantly lower levels of Ileibacterium, Faecalibaculum, and Erysipelotrichaceae_norank were observed in 5 × FAD mice treated with STZ than in the controls (Figure 2d). These findings indicate that DM can alter the intestinal flora in AD, thereby exacerbating disease progression.
3.3. Oral Administration of Butyrate Ameliorates Aβ deposition and Improves the Gut Flora of AD Mice Treated With STZ
A C4‐treated DM animal model was established as previously described in the Materials and Methods. Immunohistochemical analysis of the brain demonstrated that Aβ deposition in the brains of mice in the treatment group was significantly lower than that in the control group (Figure 3a,b). These findings suggest that butyrate exerts a protective effect against disease progression in AD mice with comorbid DM.
Figure 3. Oral administration of butyrate ameliorates Aβ deposition and improves the gut flora of AD mice treated with STZ. n = 6 for each group. (a) Immunohistochemical staining for Aβ of representative sections from brain tissues of 5 × FAD treated with STZ and C4‐treated 5 × FAD mice treated with STZ (upper: original magnification ×50; lower: original magnification ×200). (b) Bar chart of numbers of Aβ ^+^ cells in each HP view, ^∗^ p < 0.05 vs. 5 × FAD with STZ. (c). Feces of 5 × FAD with STZ treatment and C4‐treated 5 × FAD mice with STZ treatment were collected, and 16S ribosomal DNA sequencing was performed. n = 3 for each group. A heatmap of the top 20 different microbiota species between the two groups was exhibited.(a)(b)(c)
Significant alterations in gut microbiota were observed in 5 × FAD mice treated with STZ. Therefore, we further analyzed the gut microbiota of STZ‐treated 5 × FAD mice with/without C4 treatment to investigate potential changes. As shown in Figure 3c, the gut microbiota of the treatment group exhibited distinct alterations compared with that of the control group. Specifically, there was a marked increase in bacteria associated with SCFA production, including Kineothrix alysoides, Odoribacter splanchnicus, and Rikenella microfusus. Additionally, changes were noted for other beneficial bacteria, such as Bacteroides acidifaciens, Muribaculum intestinale, and Limosilactobacillus reuteri_D. Therefore, our findings suggest that butyrate may protect against AD progression by modulating gut microbiota composition.
4. Discussion
Increasing evidence suggests a strong association between DM and AD, as both conditions share pathological characteristics, including insulin resistance, impaired glucose metabolism, and mitochondrial defects [24]. However, the precise mechanisms through which DM influences cognitive decline have not yet been fully elucidated. In our experiments, diabetic AD model mice showed more severe Aβ accumulation in the brain compared with nondiabetic AD mice, suggesting that DM accelerates AD progression. Furthermore, diabetic AD mice exhibited significant intestinal barrier dysfunction. We also observed reduced MCU2 and E‐cadherin levels in the colonic tissue of these mice, indicating compromised intestinal integrity. This dysfunction likely facilitates the translocation of harmful substances into the bloodstream, which can negatively affect brain health and contribute to AD pathology. In addition to issues regarding the intestinal barrier, our analysis revealed notable changes in the gut microbiome of diabetic AD mice. Specifically, we observed a marked reduction in the number of beneficial bacteria that produce SCFAs, such as Ileibacterium and Faecalibaculum, along with an increase in harmful species like Escherichia–Shigella. SCFAs play crucial roles in maintaining gut health and influencing neuroinflammation and neurodegeneration. Therefore, a C4‐treated AD mouse model was constructed, and the deposition of Aβ in the brain was examined. In the C4‐treated AD mice model, a significant decrease in Aβ accumulation was observed, whereas fecal microbial diversity analysis confirmed the beneficial effects of C4 treatment. In conclusion, our findings suggest that DM exacerbates AD through gut microbiome dysregulation and butyrate may play a beneficial role in AD progression.
Epidemiological and molecular evidence increasingly suggests a considerable overlap in the risk, comorbidities, and pathophysiological mechanisms between DM and AD. A meta‐analysis of unpublished data revealed that individuals with DM have an approximately 60% greater risk of developing dementia than those without DM, with an additional higher risk of vascular dementia in women [25]. As DM primarily affects insulin secretion and metabolism and insulin receptors are present in nearly all brain cells, insulin receptors and their related signaling pathways have garnered attention [26]. However, whether the two conditions are mechanistically linked or represent unrelated occurrences during aging remains unclear. To explore the effects of DM on AD, we constructed a classical AD mouse model using 5 × FAD mice and induced DM by injecting STZ to impair pancreatic islet function. Blood glucose monitoring and insulin staining confirmed the successful establishment of this DM model. We then examined Aβ plaque deposition in the brain tissue of 5 × FAD mice with and without STZ treatment. A significant increase in Aβ deposition was observed in the brains of 5 × FAD mice treated with STZ compared with the controls, indicating that a hyperglycemic state aggravates Aβ deposition. However, the underlying mechanism remains unknown.
The gut microbiota is profoundly involved in AD pathogenesis, influencing blood–brain barrier permeability, energy homeostasis, and synaptic transmission [27]. Germ‐free APP/PS1 and 5 × FAD mice exhibit lower Aβ levels, which can be reversed by colonizing them with the gut microbiome from conventionally raised mice [28]. This suggests that manipulating the intestinal microbiota significantly influences amyloid pathology in AD mice. Further research indicates that these effects may be achieved by modulating microglial function and the immune clearance of amyloids from the brain. Changes in microglial morphology and activation status have been observed in AD model mice whose microbiota were depleted with antibiotics; these changes were reversed by transplanting the gut microbiome from normally raised AD model mice, demonstrating the crucial link of microglia with the intestinal microbiota and amyloid pathology [29]. However, the roles of specific bacterial species in AD progression are not well established.
Considering the key role of intestinal barrier integrity in maintaining gut environmental homeostasis, we explored intestinal mucosal integrity. We observed a significant decrease in MUC2 and E‐cadherin levels in 5 × FAD mice administered STZ than in the controls, which suggested that DM damaged intestinal mucosal integrity. Microbial diversity analysis of feces from 5 × FAD mice with and without STZ treatment revealed shifts in the microbiome and increases in specific species, such as Ileibacterium, Faecalibaculum, and Ferysipelotrichaceae_norank. This change was accompanied by more pronounced Aβ deposition in the brain. Importantly, these species are closely associated with SCFA metabolism. SCFAs, including acetate, propionate, and butyrate, are key mediators in the gut–brain axis and influence the progression of neurodegenerative diseases, such as AD and Parkinson′s disease [30, 31]. Animal experiments have shown that the oral administration of acetate can modulate Aβ pathology and microglial phagocytosis rates in specific pathogen‐free mice [28]. A strong connection between SCFAs and microglia has been reported, and the introduction of B. fragilis has shown to contribute to higher levels of aggregated Aβ in the cortex of mice, attributable to polyunsaturated fatty acid metabolism [32]. These insights highlight the importance of the interaction between SCFAs and neurodegenerative diseases.
SCFAs also influence DM management by regulating the immune response. Pingitore et al. demonstrated the beneficial effects of diet‐derived SCFAs on β‐cell function in vivo and their direct influence on potentiating glucose‐stimulated insulin release in vitro [33]. Genome‐wide genotyping, gut metagenomic sequencing, and fecal SCFA level analysis of normoglycemic individuals confirmed the causal effect of the gut microbiome on metabolic traits and demonstrated that increasing the production of butyrate had beneficial effects on β‐cell function [34]. Based on our previous results, decreasing levels of microbes are closely related to reduced SCFA production in 5 × FAD mice treated with STZ, which may accelerate AD progression. Collectively, these data suggest that DM aggravates AD development by reducing microbes associated with SCFAs in the colon via the disruption of intestinal barrier integrity. Thus, SCFAs and the microbes that produce them may offer a promising avenue for AD treatment.
We established an STZ‐induced AD mouse model and treated it with C4. Immunohistochemistry revealed that Aβ deposition was significantly reduced in the brains of the C4‐treated group compared with those in the control group, which is consistent with our previous results. We collected fecal samples from both the treatment and control groups for microbiota sequencing to elucidate the underlying mechanisms. The results indicated a marked increase in SCFA‐producing bacteria following C4 treatment, including Kineothrix alysoides, Odoribacter splanchnicus, and Rikenella microfusus [35–37]. Additionally, we observed an upward trend in bacteria associated with neurodegenerative processes, such as Limosilactobacillus reuteri_D, a probiotic that has been shown to modulate AD progression [38]. Certain immune‐related bacteria were also increased in the treatment group, including Bacteroides acidifaciens, which promotes gut immune homeostasis by enhancing intestinal IgA production, and Muribaculum intestinale, known to stimulate IL‐10 secretion and exert anti‐inflammatory effects [39, 40]. These findings are consistent with our previous results, suggesting that enhancing the abundance of SCFA‐producing bacteria in the gut could help restore intestinal integrity, reduce systemic inflammation, and protect against cognitive decline.
5. Conclusions
Despite limitations, such as the lack of metabolomics analyses and additional investigation of bacterial communities (e.g., fecal microbiota transplantation), our results provide new insights into how DM facilitates Aβ plaque deposition in the brain through intestinal barrier damage and gut microbiome alterations. These findings enhance our understanding of AD pathogenesis and identify intestinal flora and butyrate supplementation as promising targets for AD treatment.
Author Contributions
Qiong He and Zixiao Zhao contributed equally to this work.
Funding
The study was funded by the National Natural Science Foundation of China, 82001143 and 82071962.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting Information Additional supporting information can be found online in the Supporting Information section. Table S1: List of the sequence information of the forward and reverse primers used for quantitative real‐time PCR (qPCR). The target genes include mouse Gapdh (glyceraldehyde‐3‐phosphate dehydrogenase), E‐cadherin, and Muc2 (mucin 2). All primers were synthesized by Sangon Biotech.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Twarowski B. and Herbet M. , Inflammatory Processes in Alzheimer′s Disease-Pathomechanism, Diagnosis and Treatment: A Review, International Journal of Molecular Sciences. (2023) 24, no. 7, 10.3390/ijms 24076518.PMC 1009534337047492 · doi ↗ · pubmed ↗
- 2Better M. A. , 2023 Alzheimer′s Disease Facts and Figures, Alzheimer′s Dement. (2023) 19, no. 4, 1598–1695, 10.1002/alz.13016.36918389 · doi ↗ · pubmed ↗
- 3Ma C. , Hong F. , and Yang S. , Amyloidosis in Alzheimer′s Disease: Pathogeny, Etiology, and Related Therapeutic Directions, Molecules. (2022) 27, no. 4.10.3390/molecules 27041210 PMC 887603735209007 · doi ↗ · pubmed ↗
- 4Silva M. V. F. , Loures C. M. G. , Alves L. C. V. , de Souza L. C. , Borges K. B. G. , and Carvalho M. D. G. , Alzheimer′s Disease: Risk Factors and Potentially Protective Measures, Journal of Biomedical Science. (2019) 26, no. 1, 10.1186/s 12929-019-0524-y, 2-s 2.0-85065517024.PMC 650710431072403 · doi ↗ · pubmed ↗
- 5Bellenguez C. , Charbonnier C. , Grenier-Boley B. , Quenez O. , Le Guennec K. , Nicolas G. , Chauhan G. , Wallon D. , Rousseau S. , Richard A. C. , Boland A. , Bourque G. , Munter H. M. , Olaso R. , Meyer V. , Rollin-Sillaire A. , Pasquier F. , Letenneur L. , Redon R. , Dartigues J. F. , Tzourio C. , Frebourg T. , Lathrop M. , Deleuze J. F. , Hannequin D. , Genin E. , Amouyel P. , Debette S. , Lambert J. C. , and Campion D. , Contribution · doi ↗ · pubmed ↗
- 6Jansen I. E. , Savage J. E. , Watanabe K. , Bryois J. , Williams D. M. , Steinberg S. , Sealock J. , Karlsson I. K. , Hägg S. , Athanasiu L. , Voyle N. , Proitsi P. , Witoelar A. , Stringer S. , Aarsland D. , Almdahl I. S. , Andersen F. , Bergh S. , Bettella F. , Bjornsson S. , Brækhus A. , Bråthen G. , de Leeuw C. , Desikan R. S. , Djurovic S. , Dumitrescu L. , Fladby T. , Hohman T. J. , Jonsson P. V. , Kiddle S. J. , Rongve A. , Sal · doi ↗ · pubmed ↗
- 7Hou Y. , Dan X. , Babbar M. , Wei Y. , Hasselbalch S. G. , Croteau D. L. , and Bohr V. A. , Ageing as a Risk Factor for Neurodegenerative Disease, Nature Reviews. Neurology. (2019) 15, no. 10, 565–581, 10.1038/s 41582-019-0244-7, 2-s 2.0-85072717929, 31501588.31501588 · doi ↗ · pubmed ↗
- 8Costa L. G. , Cole T. B. , Dao K. , Chang Y. C. , Coburn J. , and Garrick J. M. , Effects of Air Pollution on the Nervous System and Its Possible Role in Neurodevelopmental and Neurodegenerative Disorders, Pharmacology & Therapeutics. (2020) 210, 107523, 10.1016/j.pharmthera.2020.107523.32165138 PMC 7245732 · doi ↗ · pubmed ↗