Two complete mitochondrial genomes of the family Paradoxosomatidae (Diplopoda, Polydesmida) with phylogenetic implications
Ming Gao, Gaoji Zhang, Yingzhu Li, Xiaxi Jia, Yuting Ding, Hongyi Liu

TL;DR
This study sequenced two millipede mitochondrial genomes and used them to improve understanding of millipede evolutionary relationships.
Contribution
The study provides complete mitochondrial genomes for two millipede species and new phylogenetic insights for the group Diplopoda.
Findings
Both mitogenomes contain 37 genes on the minor strand with high AT content.
Phylogenetic analysis placed the two species in distinct clades and supported interordinal relationships.
Codon usage patterns and gene arrangement suggest a conserved molecular synapomorphy for the group.
Abstract
Millipedes (class Diplopoda) are vital soil invertebrates that play key roles in litter decomposition and nutrient cycling. However, their phylogenetic relationships remain poorly resolved due to limited genomic resources. In this study, we sequenced and characterized the complete mitochondrial genomes (mitogenomes) of two paradoxosomatid millipedes, Oxidus gracilis (15,034 bp) and Kronopolites swinhoei (15,277 bp). Both mitogenomes contain the typical set of 37 genes, all located on the minor strand (N-strand), and display high AT content. The conserved gene arrangement observed here may represent a molecular synapomorphy for this taxonomic group. Analysis of codon usage revealed that start codons includ ATN (ATA/ATG/ATT), TTG, and GTG, while stop codons consisted of TAN (TAA/TAG/TAT) and an incomplete single T. Relative synonymous codon usage (RSCU) analysis indicated that Leu2, Val,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
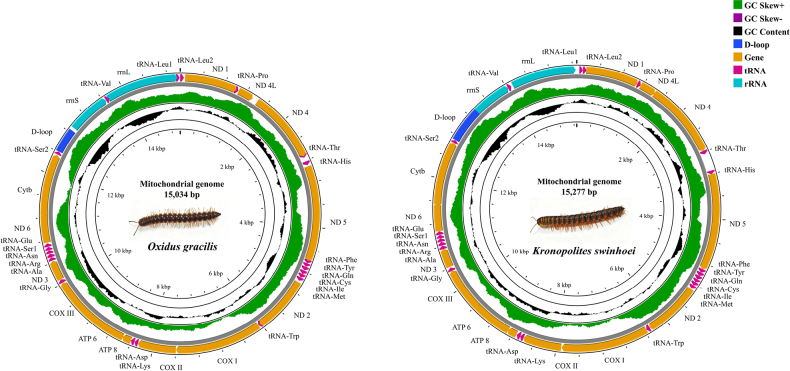 Figure 1
Figure 1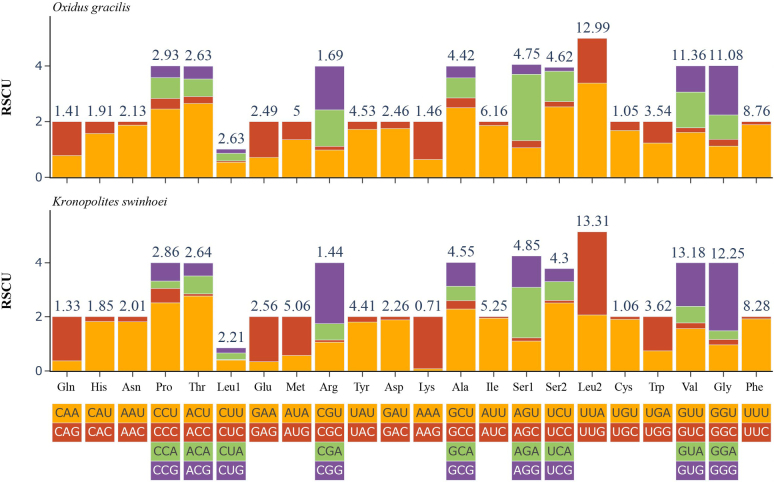 Figure 2
Figure 2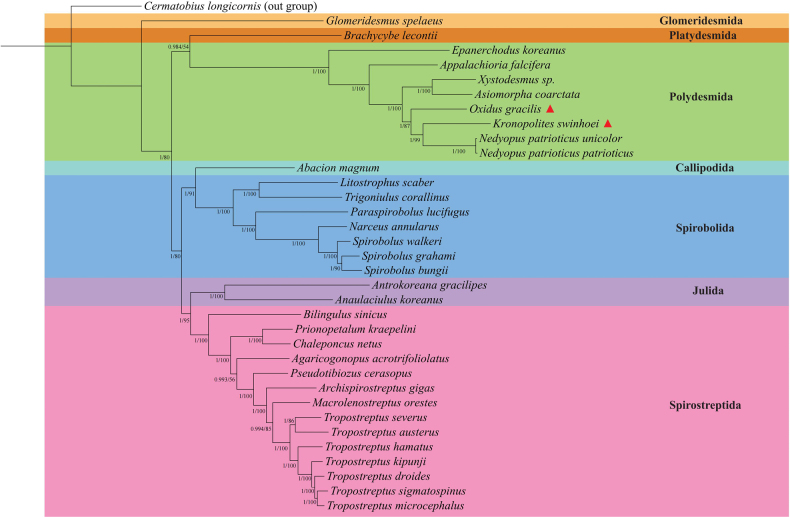 Figure 3
Figure 3| Class | Order | Family | Species | Length (bp) | Accession |
|---|---|---|---|---|---|
|
|
|
| 15,160 |
| |
|
|
| 15,115 |
| ||
|
|
| 15,282 |
| ||
| 15,791 |
| ||||
|
| 15,644 |
| |||
|
|
|
| |||
| 15,814 |
| ||||
| 15,798 |
| ||||
|
|
|
| |||
|
| 15,581 |
| |||
|
|
| 14,906 |
| ||
|
| 15,081 |
| |||
|
| 14,929 |
| |||
|
| 14,868 |
| |||
| 14,879 |
| ||||
| 14,875 |
| ||||
| 14,879 |
| ||||
|
|
| 14,747 |
| ||
|
| 14,916 |
| |||
|
|
| 15,879 |
| ||
|
| 15,114 |
| |||
| 15,093 |
| ||||
|
| 15,016 |
| |||
|
| 15,121 |
| |||
| 15,177 |
| ||||
| 15,367 |
| ||||
| 15,209 |
| ||||
| 15,261 |
| ||||
| 15,156 |
| ||||
| 15,170 |
| ||||
| 15,172 |
| ||||
| 15,176 |
| ||||
| 15,169 |
| ||||
|
|
|
| 16,833 |
|
| Gene | Location | Intergenic Nucleotides | Size (bp) | |
|---|---|---|---|---|
| From | To | |||
| tRNA-Leu2 | 1 / 109 | 66 / 171 | 10 / -1 | 66 / 63 |
| ND1 | 86 / 166 | 992 / 1104 | 19 / -6 | 907 / 939 |
| tRNA-Pro | 993 / 1098 | 1057 / 1160 | 0 / -7 | 65 / 63 |
| ND4L | 1064 / 1162 | 1339 / 1443 | 6 / 1 | 276 / 282 |
| ND4 | 1414 / 1434 | 2674 / 2771 | 74 / -10 | 1261 / 1338 |
| tRNA-Thr | 2675 / 2772 | 2741 / 2838 | 0 / 0 | 67 / 67 |
| tRNA-His | 2814 / 3140 | 2882 / 3201 | 72 / 301 | 69 / 62 |
| ND5 | 2885 / 3199 | 4583 / 4905 | 2 / -3 | 1699 / 1707 |
| tRNA-Phe | 4584 / 4905 | 4650 / 4970 | 0 / -1 | 67 / 66 |
| tRNA-Tyr | 4651 / 4970 | 4713 / 5036 | 0 / -1 | 63 / 67 |
| tRNA-Gln | 4716 / 5037 | 4781 / 5102 | 2 / 0 | 66 / 66 |
| tRNA-Cys | 4788 / 5104 | 4853 / 5164 | 6 / 1 | 66 / 61 |
| tRNA-Ile | 4855 / 5165 | 4920 / 5230 | 1 / 0 | 66 / 66 |
| tRNA-Met | 4922 / 5231 | 4985 / 5295 | 1 / 0 | 64 / 65 |
| ND2 | 5016 / 5308 | 5994 / 6303 | 30 / 12 | 1009 / 996 |
| tRNA-Trp | 5995 / 6304 | 6057 / 6369 | -30 / 0 | 63 / 66 |
| COX I | 6059 / 6371 | 7591 / 7903 | 1 / 1 | 1533 / 1533 |
| COX II | 7595 / 7913 | 8272 / 8590 | 3 / 9 | 678 / 678 |
| tRNA-Lys | 8273 / 8591 | 8336 / 8655 | 0 / 0 | 64 / 65 |
| tRNA-Asp | 8340 / 8655 | 8404 / 8720 | 3 / -1 | 65 / 66 |
| ATP8 | 8405 / 8730 | 8563 / 8894 | 0 / 9 | 159 / 165 |
| ATP6 | 8560 / 8888 | 9223 / 9592 | -4 / -7 | 664 / 705 |
| COX III | 9224 / 9555 | 10009 / 10340 | 0 / -38 | 786 / 786 |
| tRNA-Gly | 10011 / 10340 | 10075 / 10402 | 1 / -1 | 65 / 63 |
| ND3 | 10077 / 10418 | 10427 / 10753 | 1 / 15 | 351 / 336 |
| tRNA-Ala | 10436 / 10752 | 10497 / 10814 | 8 / -2 | 62 / 63 |
| tRNA-Arg | 10498 / 10818 | 10563 / 10882 | 0 / 3 | 66 / 65 |
| tRNA-Asn | 10567 / 10884 | 10632 / 10946 | 3 / 1 | 66 / 63 |
| tRNA-Ser1 | 10633 / 10947 | 10690 / 11005 | 0 / 0 | 58 / 59 |
| tRNA-Glu | 10694 / 11007 | 10755 / 11070 | 3 / 1 | 62 / 64 |
| ND6 | 10768 / 11087 | 11271 / 11560 | 12 / 16 | 504 / 474 |
| Cytb | 11225 / 11541 | 12343 / 12662 | -47 / -20 | 1119 / 1122 |
| tRNA-Ser2 | 12355 / 12663 | 12411 / 12719 | 11 / 0 | 57 / 57 |
| D-loop | 12412 / 12720 | 12848 / 13343 | 0 / 0 | 437 / 624 |
| rrnS | 12904 / 13344 | 13649 / 14025 | 55 / 0 | 746 / 682 |
| tRNA-Val | 13654 / 14023 | 13717 / 14087 | 4 / -3 | 64 / 65 |
| rrnL | 13698 / 14117 | 14983 / 15259 | -20 / 29 | 1286 / 1143 |
| tRNA-Leu1 | 14961 / 46 | 15024 / 107 | -23 / 63 | 64 / 62 |
| Species | Whole genome | PCGs | tRNAs | rRNAs | D-loop | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | AT (%) | AT skew | GC skew | Size (bp) | AT (%) | Size (bp) | AT (%) | Size (bp) | AT% | Size (bp) | AT% | |
|
| 15,034 | 66.02 | -0.24 | 0.42 | 10,916 | 64.49 | 1415 | 68.34 | 2032 | 70.13 | 437 | 73.46 |
|
| 15,277 | 60.61 | -0.32 | 0.54 | 11,061 | 59.47 | 1404 | 64.17 | 1825 | 63.89 | 624 | 70.19 |
|
| 15,644 | 67.44 | -0.26 | 0.43 | 11,021 | 66.11 | 1482 | 69.84 | 2016 | 69.35 | 954 | 75.68 |
|
| 15,282 | 64.04 | -0.37 | 0.44 | 11,009 | 63.12 | 1399 | 66.40 | 2025 | 68.74 | 880 | 60.57 |
|
| 15,581 | 75.11 | -0.26 | 0.45 | 10,965 | 73.89 | 1371 | 77.10 | 2082 | 79.06 | 411 | 69.34 |
|
| 15,814 | 68.15 | -0.26 | 0.46 | 10,904 | 66.99 | 1203 | 67.66 | 2086 | 72.63 | 1168 | 67.72 |
| 15,791 | 67.01 | -0.22 | 0.47 | 11,030 | 65.62 | 1363 | 72.05 | 2007 | 69.21 | 1032 | 70.06 | |
| Gene | Start codon / stop codon | ||||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
| ||
| ATP6 | ATA/T | ATG/TAG | ATG/T | GTG/T | GTG/T | ATA/TAA | ATG/TAA |
| ATP8 | ATG/TAG | ATG/TAG | ATG/TAA | GTG/TAG | TTG/T | ATG/TAG | ATG/TAA |
| COX I | ATG/TAA | ATG/TAG | ATG/TAG | ATG/TAA | GTG/TAA | ATG/TAG | ATG/TAG |
| COX II | ATG/TAA | ATG/TAA | ATG/TAG | ATG/T | ATG/T | ATG/TAA | ATG/TAG |
| COX III | ATG/TAA | ATG/TAA | ATG/TAA | ATA/TAG | ATG/TAG | ATG/TAA | ATG/TAG |
| Cytb | ATG/TAA | ATG/TAG | ATG/TAA | ATG/TAG | ATG/TAT | ATG/TAG | ATG/TAA |
| ND1 | ATG/T | TTG/TAG | TTG/T | GTG/T | ATG/TAG | ATA/TAT | TTG/T |
| ND2 | ATT/T | ATG/TAG | TTG/TAA | TTG/T | GTG/TAA | ATT/TAG | TTG/TAA |
| ND3 | ATG/TAG | ATT/TAG | GTG/TAA | TTG/T | ATA/TAA | ATT/TAG | GTG/TAG |
| ND4 | ATT/T | ATT/TAG | GTG/TAG | ATG/TAG | ATT/TAA | ATG/TAG | GTG/TAA |
| ND4L | ATA/TAG | ATG/TAG | ATG/TAG | ATG/TAG | ATG/T | ATG/TAG | ATG/TAG |
| ND5 | ATG/T | ATT/TAG | ATG/TAG | ATG/TAA | ATG/T | ATG/T | ATG/TAG |
| ND6 | ATA/TAA | ATT/TAA | ATG/T | TTG/T | TTG/T | ATA/TAA | ATG/T |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSubterranean biodiversity and taxonomy · Genomics and Phylogenetic Studies · Protist diversity and phylogeny
Introduction
Soil invertebrates are indispensable to ecosystem functioning, driving fundamental processes such as litter decomposition, soil formation, and nutrient cycling. They enhance the availability of key nutrients (C, N, P, K) and improve soil physical and microbial properties (Jegede et al. 2019; Wu et al. 2023; Wu et al. 2025). Millipedes, as key soil invertebrates, play a critical role in decomposing litter (particularly fallen leaves) and maintaining soil fertility (Culliney 2013). Millipedes are classified under the class of Diplopoda, with over 18,000 described species (Golovatch and Liu 2020). The majority of millipede species are currently identified primarily through morphological approaches (Mesibov 2019). However, molecular and biogeographic evidence indicates the existence of a considerable number of undescribed species, with previous estimates ranging from 15,000 to 80,000 (Sierwald and Bond 2007; Shear 2011). Morphological identification for millipedes, while historically predominant, exhibits notable limitations due to the group’s high diversity and the prevalence of homoplastic traits shaped by environmental adaptations (Liu et al. 2017).
Traditional identification methods face some challenges in identification (Engh et al. 2003). Mitochondrial genome (mitogenome) exhibits several distinctive characteristics, including strict maternal inheritance, rapid evolutionary rate, and an absence of genetic recombination (Tuli et al. 2022). Their mitochondrial DNA is characterized by a closed, circular, double-stranded configuration and undergoes semi-conservative replication (Scarpulla 2008). The mitogenome is typically composed of 37 coding elements, comprising 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes (rRNAs), and a D-loop region (Xu et al. 2024, 2025). As an ideal genetic marker, it provides a novel, even completely new, perspective for species identification, evolutionary analysis, and phylogenetic inference. Extensive reorganization of both tRNAs and PCGs was found among and within the subphyla Chelicerata, Myriapoda, and Crustacea using mitogenome (Sterling-Montealegre and Prada 2023). The mitogenomes of four millipede species were sequenced to investigate evolutionary relationships among diplopod taxa and characterize their genomic features (Li et al. 2025). Currently, complete mitogenome sequences are available for fewer than 50 millipede species, highlighting a significant gap in genomic understanding of millipedes (Li et al. 2025). A robust and comprehensive phylogenetic analysis of the class Diplopoda is contingent upon the acquisition of a substantial number of complete mitogenomes from its diverse subordinate species (Golovatch and Liu 2020).
The phylogenetic position of the family Paradoxosomatidae within the order Polydesmida remains underexplored and poorly resolved (Yang et al. 2018; Zhang et al. 2024b). Due to the scarcity of robust molecular phylogenetic frameworks for this family, the evolutionary relationships between Paradoxosomatidae and other polydesmidan lineages are still unclear. To address this gap, we sequenced and characterized the complete mitochondrial genomes of two paradoxosomatid species: Oxidus gracilis (Koch, 1847) and Kronopolites swinhoei (Pocock, 1895). Their mitogenomic analysis not only provides insights into gene rearrangement patterns within Paradoxosomatidae but also contributes to broader efforts in documenting the undersampled phylogenetic diversity of millipedes. We assembled and annotated the mitogenomes of O. gracilis and K. swinhoei and conducted a comprehensive analysis of key parameters, including genome size, nucleotide composition, codon usage, AT-skews, and GC-Skew. To resolve evolutionary relationships within Diplopoda, we employed Bayesian inference (BI) and maximum likelihood (ML) methods on these whole mitogenome data to reconstruct a robust phylogenetic framework. This study aims to provide new molecular evidence for characterizing millipede mitogenomes and to refine the phylogenetic relationships within millipedes group.
Materials and methods
Sample collection and identification
Specimens of O. gracilis and K. swinhoei were originally collected from two distinct localities in Nanjing, China: Mufu Mountain (32°07'N, 118°47'E) and Zijin Mountain (32°04'N,118°51'E). The identification of these millipedes was performed based on diagnostic morphological characteristics following established taxonomic descriptions (Iniesta et al. 2020; Xiong et al. 2025). Total genomic DNA was extracted using the DNAiso (Takara, Beijing, China) following the manufacturer’s protocol. All extracted DNA samples were archived at -80 °C in the Zoology Laboratory of Nanjing Forestry University for long-term preservation and future use.
Next-Generation sequencing, mitochondrial genome assembly, and annotation
The sequencing procedure was conducted following established Illumina next-generation sequencing protocols. Genomic DNA was initially fragmented to approximately 350 bp using a Covaris ultrasonication system, followed by end repair, A-tailing, and ligation of full-length Illumina adapters for library construction. Sequencing was performed using a PE150 paired-end strategy. Raw sequencing data underwent stringent quality control through Fastp, with filtering criteria including: (1) removal of reads containing adapter sequences (>10 bp alignment); (2) exclusion of reads with > 10% undetermined bases, and (3) elimination of reads where > 50% of bases had Phred quality scores below 5. This process yielded high-quality data for subsequent bioinformatic analyses. All procedures described above were conducted by Novogene (Nanjing, China). Geneious Prime v2024.0.7 was employed to assemble the complete mitogenome through reference-guided assembly, utilizing the published genomes of Abacion magnum and Nedyopus patrioticus as reference templates. Assembly was performed under medium sensitivity/speed parameters, yielding a consensus sequence with a 99% base-call threshold. The circular structure of the assembled mitogenome was confirmed by manual inspection of the terminal region alignments. The two mitogenomes underwent preliminary examination using Seqman v7.1.0 (Jin and Sun 2018) and were subsequently annotated with the MITOS WebServer (Meng et al. 2019) to determine the precise genomic locations of tRNAs, rRNAs, the D-loop region, and all PCGs.
Sequence analysis and phylogenetic analysis
Genomic visualization was performed by constructing a circular diagram with Proksee (Subbiah V.K. et al. 2024). Relative synonymous codon usage (RSCU) was conducted in MEGA11 (Kumar et al. 2018), and visualized with PhyloSuite v1.2.3 (Zhang et al. 2020). Nucleotide composition skewness was assessed based on established formulae: AT-skew = (A − T)/(A + T) and GC-skew = (G − C)/(G + C) (Perna and Kocher 1995).
The phylogenetic relationships among millipede groups were reconstructed based on the complete mitogenomes of 34 species, using Cermatobius longicornis (Takakuwa, 1939) as the outgroup (Table 1). The phylogenetic dataset was derived from the concatenated sequences of the complete suite of 13 PCGs of mitochondrial genes. Phylogenetic analyses for each dataset were conducted through both ML and BI using the integrated PhyloSuite platform v1.2.3 (Zhang et al. 2020; Xiang et al. 2023). Sequence alignment was performed using MAFFT v.7.313 (Rozewicki et al. 2019), and the best-fit substitution model was selected under the Bayesian Information Criterion (BIC) in ModelFinder (Kalyaanamoorthy et al. 2017). The selected models were GTR+F+R5 for ML and GTR+F+I+G4 for BI.. ML trees were reconstructed in IQ-TREE v2.2.0 (Nguyen et al. 2015) with 5,000 ultrafast bootstrap replicates (Minh et al. 2013) under the GTR+F+R5 model. BI was carried out in MrBayes v3.2.7a (Ronquist et al. 2012) with two independent Markov chain Monte Carlo (MCMC) runs of 2,000,000 generations each, sampling every 1000 steps. The first 25% of trees were discarded as burn-in after confirming convergence, as indicated by an average standard deviation of split frequencies (ASDSF) below 0.01.
Results and discussion
Mitochondrial genome organization
The complete mitogenomes of O. gracilis and K. swinhoei were characterized as circular double-stranded DNA molecules, with lengths of 15,034 bp and 15,277 bp, respectively (Fig. 1). Each assembly contained the complete set of 37 mitochondrial genes: 13 PCGs, 22 tRNAs, 2 rRNAs and a D-loop region. All genes in both mitogenomes were located on the minor strand (N-strand). Notably, this N-strand localization pattern is shared with other members of the family Paradoxosomatidae, including Nedyopus patrioticus patrioticus and Nedyopus patrioticus unicolor (Zhang et al. 2024b). This genomic arrangement was also observed across other polydesmidan lineages, suggesting that it may represent a conserved trait within the order (Dong et al. 2016; Joo et al. 2020). Gene overlaps were also identified in the mitogenomes of both millipede species. The longest intergenic overlap reached 47 bp between ND6 and Cytb in O. gracilis, whereas it extended to 38 bp between ATP6 and COX III in K. swinhoei (Table 2). Similar overlapping phenomena have been reported in other Diplopoda and soil invertebrates (Zhang et al. 2024b; Xu et al. 2025). Under evolutionary pressure toward genomic compactness, gene overlap represents an economically efficient strategy, likely reflecting selection for energy conservation and high efficiency in genetic material replication and maintenance (Doublet et al. 2015).
Mitochondrial genomes of Oxidus gracilis Koch, 1847 and Kronopolites swinhoei Pocock, 1895.
Table 2.: General features of the mitogenomes of O. gracilis and K. swinhoei.
In general, both mitogenomes exhibited highly conserved organizational features. Nucleotide composition of O. gracilis was A: 25.22%, T: 40.80%, G: 23.76%, C: 9.63%, while that of K. swinhoei was A: 20.46%, T: 40.15%, G: 30.29%, C: 9.10%. A notably high T and low C content were observed as a shared characteristic in both species. Based on nucleotide composition analyses of Polydesmida species, the genomes exhibited a marked AT bias, with values ranging from 60.61% in K. swinhoei to 75.11% in E. koreanus Verhoeff, 1937, while the corresponding G and C contents were notably low (Table 3). High overall AT content observed in the studied millipedes may represent a molecular characteristic common to invertebrates (Gissi et al. 2008). This view was further supported by the detection of a similarly elevated AT content (77.8%) in another invertebrate, Thrips hawaiiensis (Wang et al. 2021). Interestingly, all AT-skew values were negative (-0.22~-0.37) and all GC-skew were positive (0.42~0.54). These skew patterns suggested strand-specific selection and mutational pressures collectively drive the high AT content in millipede mitogenomes (Gissi et al. 2008).
PCGSs and codon usage
The combined length of PCGs was 10,916 bp in O. gracilis and 11,061 bp in K. swinhoei, accounting for 72.61% and 72.40% of their respective complete mitogenomes. Among these PCGs, ND5 was the largest and ATP8 the smallest in both species (Table 2). This pattern is also observed in other diplopods, including Agaricogonopus acrotrifoliolatus, Bilingulus sinicus, Paraspirobolus lucius, and Trigoniulus corallinus (Li et al. 2025). As shown in Fig. 1, the arrangement of all PCGs was conserved between the two species: ND1, ND4L, ND4, ND5, ND2, COX I, COX II, ATP8, ATP6, COX III, ND3 and Cytb. Due to the structural constraints imposed by the circular construction of mitogenomes, an identical arrangement of PCGs is maintained and observed in N. patrioticus (Zhang et al. 2024b). Notably, as N. patrioticus, O. gracilis, and K. swinhoei all belong to the family Paradoxosomatidae, this shared gene arrangement may serve as a diagnostic molecular information for the family.
Analysis of codon usage within Polydesmida revealed that most PCGs were found to initiate with an ATN (ATA/T/G) as start codon, while a minority utilized TTG or GTG as alternative start codons (Table 4). Spirobolus bungii was also observed to follow this pattern of start and stop codon usage (Xu et al. 2022). In O. gracilis, eight PCGs (ATP8, COX I, COX II, COX III, Cytb, ND1, ND3, ND5) started with ATG; three (ATP8, ND4L, ND6) with ATA; and two (ND2, ND4) with ATT. In K. swinhoei, eight PCGs (ATP6, ATP8, COX I, COX II, COX III, Cytb, ND2, ND4L) used ATG; four used ATT; and only ND1 used TTG. The unusual start codon TTG in ND1 was also reported in Asiomorpha coarctata and Xystodesmus sp. (Dong et al. 2016). The majority of termination codons were typical TAN codons (TAA/TAG/TAT), with a single nucleotide T notably serving as a stop codon. This abbreviated termination signal was also documented in other soil invertebrates (Zhu et al. 2018; Cheng et al. 2021). It was hypothesized that such incomplete codons were post-transcriptionally modified into complete stop codons (TAA or TAG) through polyadenylation to enable proper translation termination (Xu et al. 2025). Furthermore, these incomplete stop codons may be located adjacent to tRNAs at the 3’ ends, a configuration considered to result from post-transcriptional polyadenylation of tRNA (Lin et al. 2025). The codon usage characteristics of O. gracilis and K. swinhoei were investigated through comparative analysis of RSCU (Fig. 2). As shown in Fig. 2, both species exhibited highly similar RSCU profiles. The three most frequently used codon families encoded Leu2, Val, and Gly, whereas Gln, Cys, and Lys represented the three least utilized codon families. Codons ending with A or U showed significantly higher usage frequencies than those ending with C or G, reflecting a pronounced AT bias at the third codon position. This pattern was also reported in Litostrophus scaber (Zhang et al. 2024a).
RSCU values of two species of millipedes. Termination codon is not included.
tRNAs, rRNAs, and D-loop
The mitogenomes of both millipede species contained 22 tRNA genes. Analysis across Polydesmida members revealed total tRNA lengths ranging from 1203 bp to 1482 bp, with O. gracilis and K. swinhoei measuring 1415 bp and 1404 bp, respectively (Table 3). The AT content of these tRNAs varied between 64.17% and 77.10%, reflecting a significant AT bias consistent with the overall mitogenomic composition. Individual tRNA genes ranged from 57 bp to 69 bp in length (Table 2). Both O. gracilis and K. swinhoei shared conserved tRNA clusters, with identical gene arrangements observed in two distinct regions: the tRNA-Phe–Tyr–Gln–Cys–Ile–Met cluster and the tRNA-Ala–Arg–Asn–Ser1–Glu cluster. These specific tRNAs arrangement has also been reported in other members of Paradoxosomatidae (Zhang et al. 2024b).
Both mitogenomes contained rrnL and rrnS, separated by a tRNA-Val gene. This structural pattern, in which the two rRNAs are flanked by tRNA-Val gene, has also been reported in other Paradoxosomatidae species (Yang et al. 2018). As shown in Table 3, total rRNA length ranged from 1825 bp to 2082 bp across the examined Polydesmida. O. gracilis contained 2032 bp of rRNAs, while K. swinhoei had the minimal value at 1825 bp. These rRNAs sequences also exhibited significant AT bias, ranging from 63.89% to 79.06%, consistent with the overall base composition of the complete mitogenomes.
The D-loop region was identified between tRNA-Ser2 and rrnS in both O. gracilis and K. swinhoei. As summarized in Table 3, its length exhibited substantial variation across Polydesmida species, spanning from 411 bp to 1168 bp. A difference of 186 bp was observed specifically between O. gracilis and K. swinhoei. This divergence in length may reflect evolutionary dynamics shaping the D-loop region, analogous to those influencing PCGs and other mitogenomic components (Zhang et al. 2023). As the longest non-coding region in mitogenomes, the D-loop displayed characteristically high A-T content, ranging from 60.57% to 75.68% across the Polydesmida (Table 3), with values of 73.46% in O. gracilis and 70.19% in K. swinhoei. The AT-rich character of D-loop regions was consistent with previous reports on mitochondrial D-loop regions in other soil invertebrates (Liu et al. 2020).
Phylogenetic analyses
This study incorporated 34 diplopod species with reliably annotated mitogenomes from 15 families, using the chilopod C. longicornis as an outgroup to reconstruct the phylogenetic tree (Fig. 3). Both ML and BI analyses of the 13 PCGs produced identical topologies, with notably supported nodal values across clades. The resulting phylogeny strongly supports taxonomic relationships within the class Diplopoda. Current taxonomy places Julida, Spirostreptida, and Spirobolida within the superorder Juliformia, though their internal relationships remain contentious (Zhang et al. 2024c). In our phylogenetic analysis, Spirostreptida and Julida formed a distinct cluster, consistent with previous mitogenome studies that support a sister-group relationship between these two orders (Zuo et al. 2022; Zhang et al. 2024c; Li et al. 2025). In addition, morphological data suggest a sister-group relationship between Spirobolida and Julida (Blanke and Wesener 2014).
A phylogenetic tree was reconstructed from nucleotide sequences of 13 PCGs across 34 Diplopoda species, with C. longicornis designated as the outgroup. The tree was inferred using both Bayesian inference (BI) and maximum likelihood (ML) methods. Branch support is indicated as posterior probabilities (PP) for BI and bootstrap values (BS) for ML, presented in PP/BS format. Species newly sequenced in this study are marked with red triangles.
A key node uniting Platydesmida and Polydesmida received strong Bayesian posterior probability (PP = 0.984) but low bootstrap support (BS = 54%). As the ASDSF in the BI analysis was below 0.01, the PP value was considered more reliable for this clade (Alfaro et al. 2003). Within Polydesmida, representatives of the families Xystodesmidae and Paradoxosomatidae formed a highly supported clade (PP = 1, BS = 100%), consistent with previously reported topological structures (Zuo et al. 2022; Zhang et al. 2024b; Li et al. 2025). Although X. sp. and Appalachioria falcifera belong to the family Xystodesmidae, X. sp. clustered with A. coarctata, which belongs to Paradoxosomatidae, indicating that a precise phylogenetic placement of X. sp. warrants further thorough investigation. Within the Paradoxosomatidae clade, O. gracilis, K. swinhoei, and N. patrioticus formed a distinct group that was clearly separated from the cluster containing X. sp. and A. falcifera. Furthermore, K. swinhoei and N. patrioticus were placed in a well-supported clade (PP = 1, BS = 99%), whereas O. gracilis and K. swinhoei occupied separate, less-supported clades, providing robust molecular evidence for their taxonomic delineation. These results confirm that integrating molecular and morphological data yields more accurate classification.
Overall, the phylogenetic analyses shed light on millipede evolutionary history, biodiversity, and arthropod phylogeny. However, it is important to note that the dataset was limited to 34 species. Expanding taxonomic sampling in future sequencing efforts will be essential to resolve persisting disputes and clarify higher-level phylogenetic relationships among millipede.
Conclusion
This study sequenced the complete mitogenomes of two species from the family Paradoxosomatidae. The overall mitogenomic architecture was highly conserved between O. gracilis and K. swinhoei, a pattern also observed in other members of Paradoxosomatidae. Notably, all genes were located on the N-strand, suggesting this may be a molecular synapomorphy for order Polydesmida. PCGs also exhibited a conserved arrangement, further supporting the notion of a characteristic genomic signature for the family Paradoxosomatidae. Two structurally conserved tRNA clusters were identified, and the rRNAs were separated by tRNA-Val. The D-loop region was located between tRNA-Ser2 and rrnS in both species. Consistent with other invertebrate mitogenomes, all genomic features displayed pronounced AT bias. Phylogenetic analyses based on these data offer new insights into millipede evolutionary history. However, the limited taxonomic sampling (34 species) constrains broader inferences. Expanding both taxon sampling and genomic data in future studies will be essential for resolving higher-level phylogenetic relationships within Diplopoda.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alfaro ME, Zoller S, Lutzoni F (2003) Bayes or bootstrap? A simulation study comparing the performance of Bayesian Markov chain Monte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Molecular Biology and Evolution 20(2): 255–266. 10.1093/molbev/msg 02812598693 · doi ↗ · pubmed ↗
- 2Blanke A, Wesener T (2014) Revival of forgotten characters and modern imaging techniques help to produce a robust phylogeny of the Diplopoda (Arthropoda, Myriapoda). Arthropod Structure & Development 43(1): 63–75. 10.1016/j.asd.2013.10.00324184600 · doi ↗ · pubmed ↗
- 3Cheng GQ, Du YM, Liu XJ (2021) Phylogenetic relationship and characterization of the complete mitochondrial genome of Cheilomenes sexmaculata (Coleoptera: Coccinellidae). Mitochondrial DNA, Part B, Resources 6(8): 2449–2451. 10.1080/23802359.2021.1955767 PMC 831258534368443 · doi ↗ · pubmed ↗
- 4Culliney TW (2013) Role of arthropods in maintaining soil fertility. Agriculture 3(4): 629–659. 10.3390/agriculture 3040629 · doi ↗
- 5Dong Y, Zhu LX, Bai Y, Ou YY, Wang CB (2016) Complete mitochondrial genomes of two flat-backed millipedes by next-generation sequencing (Diplopoda, Polydesmida). Zoo Keys 637: 1–20. 10.3897/zookeys.637.9909 PMC 524011828138271 · doi ↗ · pubmed ↗
- 6Doublet V, Ubrig E, Alioua A, Bouchon D, Marcadé I, Maréchal-Drouard L (2015) Large gene overlaps and t RNA processing in the compact mitochondrial genome of the crustacean Armadillidium vulgare. RNA Biology 12(10): 1159–1168. 10.1080/15476286.2015.1090078 PMC 482927126361137 · doi ↗ · pubmed ↗
- 7Engh AL, Nelson KG, Peebles R, Hernandez AD, Hubbard KK, Holekamp KE (2003) Coprologic survey of parasites of spotted hyenas (Crocuta crocuta) in the Masai Mara National Reserve, Kenya. Journal of Wildlife Diseases 39(1): 224–227. 10.7589/0090-3558-39.1.22412685088 · doi ↗ · pubmed ↗
- 8Gissi C, Iannelli F, Pesole G (2008) Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101(4): 301–320. 10.1038/hdy.2008.6218612321 · doi ↗ · pubmed ↗