Case Report: Whole-genome sequencing of urothelial carcinoma in an adult patient with CLOVES syndrome reveals a lack of PIK3CA mutation and a genomic landscape consistent with urothelial carcinoma
Lauren McAuley, Orla Fitzpatrick, Nicola Cosgrove, Jad Yacoub, Liam Grogan, Bryan T. Hennessy, Simon J. Furney, Sinead Toomey

TL;DR
Whole-genome sequencing of a urothelial carcinoma in a patient with CLOVES syndrome found no PIK3CA mutations, indicating the cancer was unrelated to the syndrome.
Contribution
This case report provides genomic insights into urothelial carcinoma in a CLOVES syndrome patient, showing no PIK3CA mutations.
Findings
Somatic alterations in the tumor were consistent with typical urothelial carcinoma features.
No somatic PIK3CA mutations were detected in the tumor tissue.
Homozygous deletions of CDKN2A and CDKN2B were observed.
Abstract
Congenital lipomatous overgrowth, vascular epidermal nevi, and skeletal abnormalities (CLOVES) syndrome is a rare genetic disorder caused by somatic activating mutations in the PIK3CA gene that arise during embryonic development. Mutations in the PI3K–AKT–mTOR pathway have been linked to various benign overgrowth disorders, including syndromes within the PIK3CA-related overgrowth spectrum. Somatic PIK3CA mutations also occur frequently across many cancer types; however, evidence linking CLOVES syndrome to increased cancer risk is not conclusive. Here, we describe a whole-genome sequencing (WGS) study of a primary pT3 high-grade urothelial carcinoma in a 62-year-old male patient diagnosed with CLOVES syndrome. A left laparoscopic nephroureterectomy was completed. Tumour tissue and a matched blood sample were collected for whole-genome sequencing, and somatic variant detection was…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
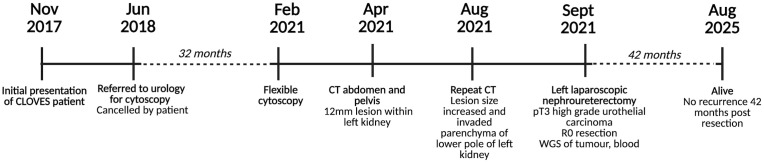 Figure 1
Figure 1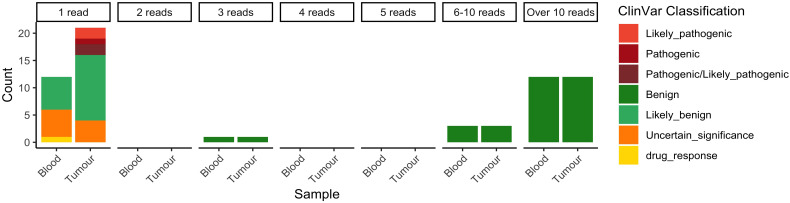 Figure 2
Figure 2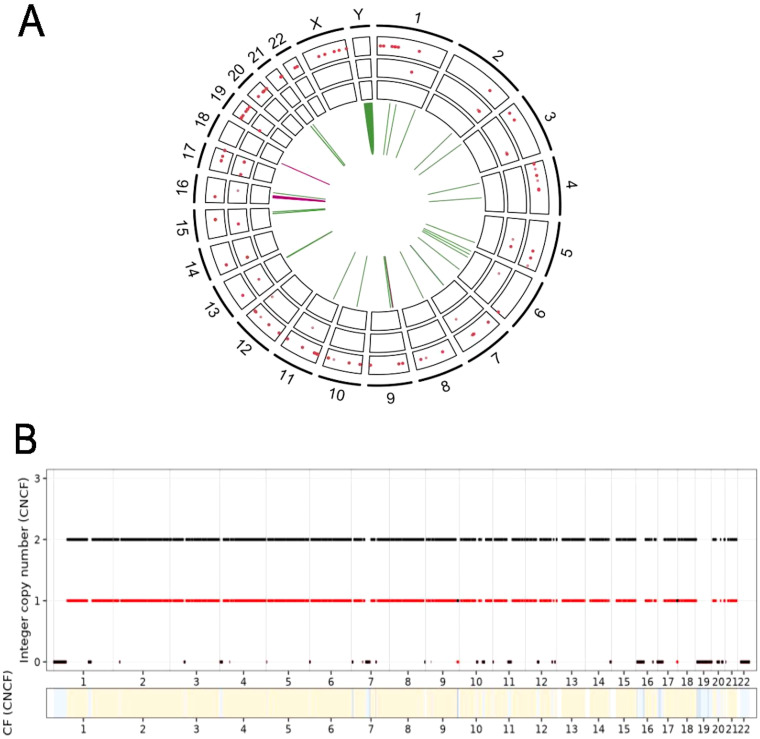 Figure 3
Figure 3| ClinVar variation ID | HGVSp | Classification | Associated conditions | Supporting reads/total reads (VAF) |
|---|---|---|---|---|
| 280875 | ENSP00000263967.3:p.Arg108His | Pathogenic/likely pathogenic | MCAP syndrome | 1/41 (2.4%) |
| 376049 | ENSP00000263967.3:p.Arg88Gln | Pathogenic | MCAP syndrome | 1/43 (2.3%) |
| 809567 | ENSP00000263967.3:p.Gly364Arg | Pathogenic/likely pathogenic | MCAP syndrome | 1/51 (1.9%) |
| 376243 | ENSP00000263967.3:p.Pro539Arg | Likely pathogenic | MCAP syndrome | 1/39 (2.5%) |
| 3774497 | ENSP00000263967.3:p.Gln546Leu | Likely pathogenic | PROS | 1/36 (2.7%) |
| SV type | Chromosome | Start position | End position | Size (bp) | VAF | Genes affected |
|---|---|---|---|---|---|---|
| INV | chr1 | 107889975 | 107890614 | 639 | 16% |
|
| INV | chr5 | 161523567 | 161524516 | 949 | 19% |
|
| INV | chr6 | 28507581 | 28508810 | 1,229 | 20% |
|
| INV | chr6 | 47964006 | 47964686 | 680 | 9% |
|
| INV | chr6 | 132448638 | 132449203 | 565 | 12% |
|
| INV | chr8 | 14192656 | 14193735 | 1079 | 10% |
|
| DEL | chr9 | 21908255 | 22712570 | 804,315 | 12% | |
| INV | chr9 | 36076171 | 36076902 | 731 | 24% |
|
| INV | chr10 | 60045041 | 60045975 | 934 | 17% |
|
| INV | chr13 | 77927036 | 77927857 | 821 | 12% |
|
| INV | chr15 | 71034937 | 71035459 | 522 | 21% |
|
| INV | chrY | 11326459 | 56843845 | 45,517,386 | 2% |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer and Skin Lesions · Renal cell carcinoma treatment · Genetic and rare skin diseases.
Background
Congenital lipomatous overgrowth, vascular epidermal nevi, and skeletal abnormalities (CLOVES) syndrome is a rare genetic disorder that is characterised by lipomatous overgrowth, vascular malformations, and skeletal abnormalities. CLOVES syndrome is caused by somatic activating mutations in the PIK3CA gene that arise during embryonic development. Thus, PIK3CA mutations in CLOVES syndrome are mosaic in nature.
PIK3CA mutations are well-known drivers of oncogenesis. PIK3CA encodes p100α, the catalytic subunit of the phosphoinositide 3-kinase (PI3K) enzyme. PI3K activates signalling through the AKT–mTOR axis to promote cell growth, metabolism, and cellular survival. Hyperactivating mutations in PIK3CA occur in approximately 11% of all cancers, with several hotspot mutations in exons 9 (E542K and E545K) and 20 (H1047R and H1047L) frequently observed (1). The mutational landscape of PIK3CA in CLOVES syndrome shares some similarities with that of cancer. Both hyperactivating hotspot PIK3CA mutations and non-hotspot mutations with lesser functional consequences occur in CLOVES syndrome (2, 3). In CLOVES syndrome, mutations in PIK3CA tend to be present at lower frequencies than in cancer due to the mosaic nature of the disorder. The genotype–phenotype relationship between hotspot/non-hotspot PIK3CA mutations and the clinical severity of CLOVES syndrome remains unclear, as the variant allele frequency (VAF) of PIK3CA mutations in CLOVES syndrome does not correlate with clinical severity, although this may be attributable to difficulties related to clinical sampling (4). Despite the link between PIK3CA and oncogenesis, there is limited evidence to suggest a higher risk of *PIK3CA-*driven cancers in patients with CLOVES syndrome (3, 5–7). Due to the rarity of CLOVES syndrome, further investigation is required to conclusively determine the cancer risk in this patient population. Here, we add to the literature a whole-genome sequencing (WGS) study of a primary urothelial carcinoma in a 62-year-old patient diagnosed with CLOVES syndrome.
Case presentation
A 62-year-old Irish male patient with a clinical diagnosis of CLOVES syndrome initially presented in November 2017 with kyphosis, pectus excavatum, and unilateral pain, with numbness and atrophy of paraspinal muscles. In 2020, genetic testing was performed, but no causative mutation was identified. A molecular diagnosis of CLOVES syndrome could not be made. A repeat biopsy was scheduled with a clinical geneticist, but the patient did not attend. Electromyography and nerve conduction studies confirmed radiculopathy, and a subsequent MRI revealed lipomatous infiltrate adjacent to the erector spinae, spinal canal stenosis, and severe atrophy of spinal discs. This was associated with significant lower limb weakness, resulting in reduced mobility and severe pain requiring specialised pain team reviews. Large capillary vascular malformations were visible on clinical examination on his right flank. The patient had previously undergone lipoma excision multiple times within a different health system.
In June 2018, the patient was referred to urology due to difficulty voiding and intermittent haematuria, persistent for 2 years. A cystoscopy was planned but cancelled by the patient. In February 2021, a flexible cystoscopy was performed after the patient presented with worsening lower urinary tract symptoms. Two months later (April 2021), CT imaging of the abdomen and pelvis identified a 12-mm lesion within the left kidney, which was radiologically concerning for urothelial carcinoma. On a repeat CT performed in August 2021, the lesion had increased in size and was now invading the parenchyma of the lower pole of the left kidney. A flexible ureteroscopy and biopsy of the lesion identified a high-grade urothelial carcinoma. In September 2021, a left laparoscopic nephroureterectomy was performed. The tumour was diagnosed as pT3 high-grade urothelial carcinoma within the renal pelvis with negative margins (R0 resection). Tumour and matched blood were collected for whole-genome sequencing. The patient elected to forgo adjuvant systemic therapy due to the risk of peripheral neuropathy associated with platinum-based chemotherapy, given his background of significant neurological deficits related to his spinal condition. Since his resection of this high-grade urothelial cancer, this patient continues with radiological, clinical, and cystoscopy follow-ups at regular intervals. He is alive and without evidence of disease recurrence 42 months post-resection. The timeline of this case is shown in Figure 1.
Timeline of this patient’s case.
Results
Single-region bulk whole-genome sequencing was performed for the resected tumour and matched blood with average coverages of 33× and 34×, respectively. A panel of 49 genes in which germline variants are known to increase urothelial carcinoma (UC) risk were assessed for germline variants in both the blood and tumour (8). Additionally, PIK3CA was also investigated for germline variants. No known pathogenic germline single-nucleotide variants (SNVs) or insertions and deletions (indels) were detected in any of these 50 genes or the associated promoters and enhancers. Additionally, no germline structural variants (SVs) or copy number variants (CNVs) affecting the 50 genes were detected. With no genetic predispositions to UC detected in the gene panel, tumour-normal analyses were performed to identify somatic SNVs, indels, multinucleotide variants (MNVs), SVs, and copy number alterations (CNAs). The details of WGS and germline/somatic variant analyses are available in the Supplementary Material.
No hyperactivating somatic mutations affecting PIK3CA were detected
Using Mutect2 and Strelka, a total of 12,691 somatic small mutations were detected genome-wide (7,760 SNVs, 4,136 indels, and 795 MNVs). Of these, just one MNV (chr3:179207972:CC: GT) affected the PIK3CA gene. This double substitution is intronic and was not predicted to affect splicing or protein function using SpliceAI. No other somatic small variants affecting the PIK3CA gene, its promoter, or associated enhancers were detected using Mutect or Strelka. Force-calling of hotspot positions at E452, E545, H1047, and N345 of PIK3CA was performed using Mutect2 in tumour-only mode on both the tumour and blood samples separately in order to prevent the exclusion of any somatic mosaic variants shared between these samples. No hotspot PIK3CA mutations were detected in the tumour or in the blood samples. Visual inspection confirmed that no activating mutations were present at hotspot positions in PIK3CA in the tumour or blood (Supplementary Figure 1).
To ensure that algorithmic limitations did not exclude any potential low-frequency small somatic variants, counts of nucleotides across the PIK3CA gene were analysed. Across the 90,754 nucleotides in the PIK3CA gene, 3,651 and 2,789 alternate alleles were supported by at least one read in the blood and the tumour samples, respectively. The majority of the alternate alleles were not classified in ClinVar; 19/2,789 and 28/3,651 alternate alleles detected in the blood and tumour, respectively, had available ClinVar classifications. No pathogenic or likely pathogenic alternate alleles were supported in the blood, while five pathogenic or likely pathogenic alternate alleles were supported in the tumour (Figure 2). While five of these alleles were previously linked to PIK3CA-related overgrowth spectrum and/or megalencephaly capillary malformation polymicrogyria (MCAP) syndrome (Table 1), the pathogenic alternate alleles here were likely to be sequencing errors, as they were supported only by single reads.
Alternate alleles reported by VarScan in blood and tumour samples with ClinVar classifications available.
No somatic SNVs or indels that may activate PIK3CA were reliably detected. No somatic structural variants affecting the PIK3CA gene, or its promoter or associated enhancers, were detected in this analysis. Somatic copy number analysis reported PIK3CA as diploid. Overall, no somatic SNVs, indels, structural variants, or copy number alterations that are known to activate PIK3CA were detected.
The mutational landscape of the tumour is consistent with that of previously reported urothelial carcinomas
Of the 12,691 somatic small mutations detected genome-wide, one driver mutation was identified. An SNV in MAP2K1 resulting in a gain-of-function mutation in the MEK1 protein (P124L) was detected at a variant allele frequency of 17%. MEK1 is an activator of the MAPK/ERK pathway. Structural variant analysis detected 33 SVs (28 inversions, four deletions, and one duplication) (Figure 3A). Of all SVs, 12 affected one or more protein-coding genes (Table 2). A large 804-kb deletion including CDKN2A and CDKN2B was detected with an estimated variant allele frequency (VAF) of 12%. Somatic copy number alteration (sCNA) analysis confirmed a homozygous loss within chromosome 9p containing CDKN2A and CDKN2B present at a frequency of 21% in the tumour sample (Figure 3B). CDKN2A and CDKN2B are tumour suppressor genes, with CDKN2A deleted in up to 42% of urothelial carcinoma cases, with CDKN2A/B deletions co-occurring in 23% of cases (9, 10). Additional homozygous deletions within chromosomes 3p, 17p, 19p, 19q, and 22q were detected, resulting in losses in multiple genes that are also recurrently altered in urothelial carcinoma, including ERCC2, CREBBP, KMT2D, PRMD4, and SETD2 (9, 10). Heterozygous loss of TSC1 was also observed. No gains were detected (Figure 3B). CREBBP, KMT2D, and SETD2 are chromatin remodelling genes, while PRMD4 and TSC1 are both tumour suppressors that regulate the PI3K pathway (11, 12). Overall, the small somatic driver mutation detected in MAP2K1 in this tumour is a rarer occurrence in urothelial carcinoma cases, while the somatic copy number profile is consistent with previous findings in urothelial carcinoma.
(A) Small non-synonymous somatic variants and structural variants detected genome-wide. SNVs are in the outer ring, indels in the middle ring, and MNVs in the inner ring. Structural variants are in the centre (green, inversions; pink, deletions; blue, duplications). (B) Somatic copy number alterations (black, total copy number; red, minor copy number; CF, cellular fraction). SNVs, single-nucleotide variants; indels, insertions and deletions; MNVs, multinucleotide variants.
Discussion
Mutations in the PI3K–AKT–mTOR pathway have been linked to benign overgrowth disorders, including hemimegalencephaly and syndromes belonging to the PIK3CA-related overgrowth spectrum (PROS), such as the Klippel–Trenaunay syndrome and CLOVES syndrome (13, 14). Somatic PIK3CA mutations frequently occur across many cancer types; however, evidence linking CLOVES syndrome to increased cancer risk is not conclusive. In a study of 122 patients, it was found that patients with CLOVES syndrome are at significantly higher risk of developing Wilms tumour, which is an embryonic renal cancer (incidence 3.3% in CLOVES vs. 0.0001% in the general population); however, the PIK3CA mutational status of these tumours was not reported (15). In another study of 267 PROS patients, six (2.2%) patients presented with cancer, including two paediatric and four adult cancers. Ultradeep sequencing was performed on 4/6 tumours; PIK3CA mutations were confirmed in one paediatric and one adult tumour (7). The relationship between CLOVES syndrome and cancer risk remains unclear. Here, whole-genome sequencing could not confirm the presence of a PIK3CA mutation in this patient’s primary urothelial carcinoma, nor were regulatory or structural alterations affecting this gene observed. Rather, a gain-of-function driver mutation in MAP2K1 was detected, which would promote signalling through the MAPK/ERK pathway via hyperactivation of MEK1. Mutations in MAP2K1 are rare across bladder cancers, occurring in 0.5% of bladder cancer samples in The Cancer Genome Atlas database. However, the regulation of the PI3K pathway is nonetheless disrupted in this tumour due to losses in PRMD4 and TSC1. Losses of these genes are common in urothelial carcinoma, as are homozygous deletions of CDKN2A and CDKN2B, both of which were also detected in this tumour. As sCNAs recurrently altered in urothelial carcinoma were detected in this tumour, and no PIK3CA activating mutations were detected, this tumour may have arisen independently of CLOVES syndrome.
A limitation of this study was the availability of only a single-region, low-purity tumour sample for genomic analyses. Intra-tumoural heterogeneity could not be investigated in this study, as multi-region samples of the tumour were not available. Average WGS coverages of 33× and 34× were achieved for tumour and blood samples, respectively, which may limit the ability to detect PIK3CA-mutated subclones. Potentially, ultradeep sequencing could have uncovered low-frequency PIK3CA mutations in this patient’s blood or tumour. Moreover, contamination of the tumour sample with normal tissues may have limited the ability to detect low-frequency somatic mutations (16). However, as PIK3CA mutations are hypothesised to be a potential driver of this patient’s tumour, such a mutation was hypothesised to be present in the tumour at a frequency detectable by the WGS data generated here.
A potential explanation for an unchanged cancer risk in CLOVES syndrome patients is that the tissue distribution of PIK3CA mutations in CLOVES syndrome differs from that of cancers carrying PIK3CA mutations. Overgrowths in CLOVES syndrome predominantly occur in tissues of mesodermal and neuroectodermal origins, while PIK3CA-mutated cancers occur in tissues derived from ectodermal and endodermal tissues (3). Alternatively, it has been proposed that PIK3CA mutations alone are not adequate tumour-initiating events (3). There is evidence to suggest that this may be the case in urothelial carcinoma; in murine models of bladder cancer, increased urothelial thickness and nuclear atypia were observed in PIK3CA^H1047^ mice, but this did not progress to invasive disease, indicating that additional genetic alterations are necessary for tumourigenesis in this context (17). While we did not detect gain-of-function mutations in PIK3CA in this case, we did observe other alterations that are characteristic of urothelial carcinoma, such as CDKN2A/B deletions and heterozygous loss of TSC1. Further investigation is required to conclusively determine cancer risk in patients with CLOVES syndrome.
Conclusion
In this case report, the whole-genome sequencing of a urothelial carcinoma diagnosed in a patient with CLOVES syndrome identified somatic variants consistent with those of previously reported urothelial carcinoma cases. No strong evidence of activating mutations affecting PIK3CA was detected in the tumour, which suggests that this patient’s urothelial carcinoma was not driven by somatic mosaic PIK3CA mutations related to CLOVES syndrome. However, multi-region sampling and/or ultradeep sequencing would be required to ensure that PIK3CA-mutated subclones do not remain undetected and that low-frequency PIK3CA variants are thoroughly investigated.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tharin Z Richard C Derangère V Ilie A Arnould L Ghiringhelli F . PIK 3CA and PIK 3R 1 tumor mutational landscape in a pan-cancer patient cohort and its association with pathway activation and treatment efficacy. Sci Rep. (2023) 13:1–12. doi: 10.1038/s 41598-023-31593-w, PMID: 36934165 PMC 10024711 · doi ↗ · pubmed ↗
- 2de Kock L Cuillerier A Gillespie M Couse M Hartley T Mears W . Molecular characterization of 13 patients with PIK 3CA-related overgrowth spectrum using a targeted deep sequencing approach. Am J Med Genet A. (2024) 194:e 63466. doi: 10.1002/ajmg.a.63466, PMID: 37949664 · doi ↗ · pubmed ↗
- 3Madsen RR Vanhaesebroeck B Semple RK . Cancer-associated PIK 3CA mutations in overgrowth disorders. Trends Mol Med. (2018) 24:856–70. doi: 10.1016/j.molmed.2018.08.003, PMID: 30197175 PMC 6185869 · doi ↗ · pubmed ↗
- 4Keppler-Noreuil KM Rios JJ Parker VER Semple RK Lindhurst MJ Sapp JC . PIK 3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. (2014) 0:287. doi: 10.1002/AJMG.A.36836, PMID: 25557259 PMC 4480633 · doi ↗ · pubmed ↗
- 5Girgis M Benedetti DJ . A case of high-risk neuroblastoma in a child with CLOVES syndrome. Pediatr Blood Cancer. (2023) 70:e 30393. doi: 10.1002/pbc.30393, PMID: 37092956 · doi ↗ · pubmed ↗
- 6Karpathiou G Chauleur C Picot T Peoc’h M . Molecular analysis of a uterine broad ligament leiomyoma in a patient with CLOVES syndrome. Pathol Res Pract. (2020) 216:153285. doi: 10.1016/j.prp.2020.153285, PMID: 33190013 · doi ↗ · pubmed ↗
- 7Faivre L Crépin JC Réda M Nambot S Carmignac V Abadie C . Low risk of embryonic and other cancers in PIK 3CA-related overgrowth spectrum: Impact on screening recommendations. Clin Genet. (2023) 104:554–63. doi: 10.1111/cge.14410, PMID: 37580112 · doi ↗ · pubmed ↗
- 8Nassar AH Abou Alaiwi S Al Dubayan SH Moore N Mouw KW Kwiatkowski DJ . Prevalence of pathogenic germline cancer risk variants in high-risk urothelial carcinoma. Genetics in Medicine. (2020) 22(4):709–718. doi: 10.1038/s 41436-019-0720-x, PMID: 31844177 PMC 7118025 · doi ↗ · pubmed ↗