Novel KISS1 Gene Mutation Leading to Male Hypogonadotropic Hypogonadism
Leonie Wittner, Santosh Mahindrakar, Ali Yasin, Sandra Nicole Scheel, Wolfgang Hoeppner, Joachim Feldkamp

TL;DR
A new mutation in the KISS1 gene is found to cause male hypogonadotropic hypogonadism, with successful treatment through hormone therapy.
Contribution
A novel heterozygous KISS1 variant c.-7C>T is identified as a cause of hypogonadotropic hypogonadism in two brothers.
Findings
The KISS1 mutation affects the Kozak sequence and may disrupt gene expression.
Testosterone therapy restored sex characteristics and hormone levels in both brothers.
Combination hormone therapy enabled spermatogenesis and fatherhood in one patient.
Abstract
The human KISS1 gene encodes the hypothalamic Kisspeptin, which is released in a pulsatile manner and binds the KISS1 receptor, that is located on gonadotropin releasing hormone neurons. This interaction ensures pulsatile gonadotropin releasing hormone secretion leading to induction of the hypothalamic-pituitary-gonadal axis and by this controls puberty onset. Disruption of this process is associated with hypogonadotropic hypogonadism. We identified a novel heterozygous KISS1 variant c.-7C>T in two brothers diagnosed with hypogonadotropic hypogonadism. The mutation affects the Kozak consensus sequence of the KISS1 gene and potentially interferes with KISS1 gene expression. Consequently, this affects the hypothalamic-pituitary-gonadal axis resulting in hypogonadotropic hypogonadism. In both patients, complete development of primary and secondary male sex characteristics and stabilization…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
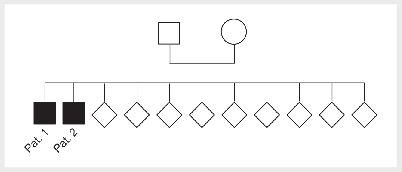 Figure 1
Figure 1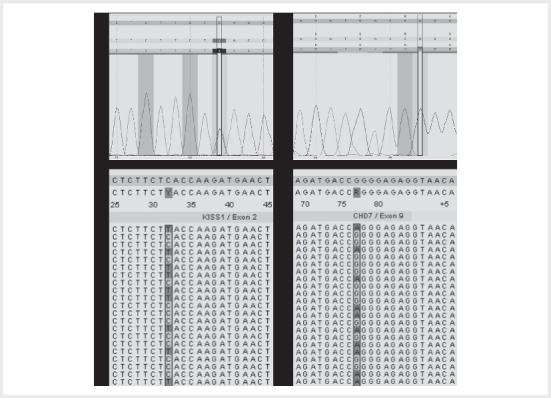 Figure 2
Figure 2| Parameter | Result | Reference |
|---|---|---|
| Volume ejaculated | 5.4 ml | > 1.5 ml |
| Time to liquefication | 10 min | < 30 min |
| Colour | Milky-white | Milky-white |
| Total sperm count | 313.2 mil | > 39 mil |
| Concentration | 58 mil/ml | > 15 mil/ml |
| Consistence | Normal | Fluid |
| pH | 8.5 | > 7.2 |
|
|
|
|
| Rapid progressive | 0 | > 32 |
| Slowly progressive | 72.7 | |
| Local progressive | 2.3 | - |
| Immotile | 25 | - |
|
|
|
|
| Normal | 2.8 | > 4 |
| Abnormal heads | 83 | - |
| Abnormal midpieces | 14.2 | - |
| Abnormal principal pieces | 0 | - |
| Parameter | Hormone serum concentration | Reference | ||
|---|---|---|---|---|
| before testosterone therapy | 3 months after therapy initiation | 10 months after therapy initiation | ||
| FSH | 1.1 mU/ml | < 0.5 mU/ml | < 0.5 mU/ml | 0.7–11.1 mU/ml |
| LH | 0.4 mU/ml | 0.4 mU/ml | < 0.4 mU/ml | 0.8–7.6 mU/ml |
| Estradiol | < 20 pg/ml | 31.2 pg/ml | 29.4 pg/ml | < 56 pg/ml |
| Testosterone | < 20 ng/dl | 384 ng/dl | 390 ng/dl | 160–726 ng/dl |
| Prolactin | 3.7 ng/ml | 15.5 ng/ml | n. a. | 2.5–17 ng/ml |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHypothalamic control of reproductive hormones · Regulation of Appetite and Obesity · Neuroendocrine regulation and behavior
Introduction
Human reproduction is tightly linked to the hypothalamic-pituitary axis 1 . Gonadotropin releasing hormone (GnRH) neurons exhibit pulsatile activity and are key drivers for the induction of puberty and human reproduction 2 3 . GnRH deficiency is an oligogenic disease and exhibits a heterogeneous clinical presentation 4 5 . It can result in hypogonadotropic hypogonadism (HH), which is characterized by low gonadotropins and sex steroid hormone levels. Ultimately, HH prevents puberty onset and is associated with infertility 5 .
Pulsatile Kisspeptin 1 (Kp) activity induces GnRH secretion that activates the hypothalamic-pituitary-gonadal axis 6 . Interfered Kp signaling is associated with HH and lack of puberty onset 7 8 . Loss of function mutations of the human KISS1 and KISS1R (KISS1 receptor) genes have been reported to cause normosmic and anosmic GnRH deficiency 7 8 , which frequently manifested in HH 9 10 11 12 13 14 15 16 .
Here, we report a novel KISS1 mutation in two brothers leading to hypogonadotropic hypogonadism.
Materials and methods
Patient consent
Written informed consent was obtained from both patients.
Next generation sequencing
DNA of both patients was isolated from whole blood samples using the QIAamp DNA blood Mini Kit (Qiagen, Hilden, Germany) according to manufacturer’s guidelines and next generation sequencing (NGS) was performed. Further clinically relevant deviations from the reference sequence apart from KISS1 and CHD7 gene mutations, were excluded with>98% probability.
Multiplex ligation-dependent probe amplification
Deletions and duplications of the ANOS1 , FGFR1 , PROK2 , PROKR2, CHD7 , GNRH1, GNRHR , and KISS1R genes were analyzed via Multiplex ligation-dependent probe amplification (MLPA). DNA was isolated as described above. MLPA was performed using SALSA MLPA Kit P050-B2 CAH (MRC-Holland) according to manufacturer’s guidelines.
Results
Clinical presentation and management
Patient 1 is the older brother and was diagnosed at the age of 29 with idiopathic HH (normoosmic). HH was pronounced with luteinizing hormone (LH) serum levels below the detection limit and testosterone serum levels below the reference values. In line with this, he presented with absent development of male sex characteristics and infertility. Testosterone therapy successfully initiated puberty resulting in complete development of primary and secondary male sex characteristics and stabilization of serum sex steroid hormone levels. Later, he received a combination therapy of human chorionic gonadotropin (hCG) and FSH which induced spermatogenesis. In response to that, his spermiogram corresponded to the normal range regarding volume, time to liquefication, colour, total sperm count, concentration, consistence, pH, and motility. However, sperm morphology presented with a high proportion of abnormal heads and midpieces (teratozoospermia) ( Table 1 ). Ultrasound examination of the testes showed normal tissue and volume with a tendency towards the lower limit range (right 9.9 ml, left 10.9 ml). Patient 1 fathered two children.
: Table 1 Spermiogram of patient 1 after hCG and FSH combination therapy.
Patient 2 is the younger brother. He presented at age 32 with a prepubescent appearance and was diagnosed with idiopathic HH: height 181 cm, arm span 190 cm, weight 90.8 kg, high voice, lack of male facial and body hair, sparse pubic hair (Tanner stage II), childlike penis (Tanner stage II). Ultrasound examination of the testes demonstrated low volume (right 2 ml, left 1.5 ml). The left testicle is palpable in the inguinal canal. X-ray examination of the left hand determined a bone age of 18 years. Hormone serum concentrations were below or at the lower reference limit ( Table 2 ). Patient 2 was also diagnosed with idiopathic HH (normoosmic). Likewise, testosterone therapy successfully induced complete development of primary and secondary male sex characteristics and stabilization of serum sex steroid hormone levels ( Table 2 ).
: Table 2 Hormone serum concentration of patient 2 undergoing testosterone therapy.
The two patients came from Iraq to Germany. Here, they received medical treatment for their symptoms for the first time, whereupon they were diagnosed with HH. For this reason, the disease was diagnosed delayed.
Further siblings of patient 1 and 2 were not affected by idiopathic HH ( Figure 1 ).
Family tree of patient 1 and 2. Patient 1 and 2 are the only siblings affected by idiopathic HH in their family.
Identification of genetic aberrations
Due to the increased familial prevalence of idiopathic HH, we screened patient 1 and 2 for genetic aberrations associated with infertility. Whole genome analysis revealed the familial heterozygous KISS1 variant c.-7C>T (dbSNP: rs369561225, frequency: A= 0.00071 (30/42216, ExAC)) in both patients ( Figure 2 a, b ). The mutation affects the mRNA 5’-untranslated region (UTR), where it is localized in the Kozak consensus sequence. It has been described earlier in a patient with Kallmann syndrome 17 .
Identification of KISS1 variant c.-7C>T and of CHD7 variant c.2690G>A. a Sequence analysis of patient 1 and b sequence analysis of patient 2 indicate the heterozygous substitution C> T at position -7 of the KISS1 gene. c Sequence analysis of patient 1 revealed no genetic aberration in CHD7 . d Sequence analysis of patient 2 indicates the heterozygous substitution G> A at position 2690.
Furthermore, only patient 2 exhibits the heterozygous CHD7 (Chromodomain Helicase DNA Binding Protein 7) variant c.2690G>A, p.Arg897Gln (dbSNP: rs773685788, frequency: A= 0.00002 (2/83202, ExAC)) ( Figure 2 c, d ). Both chromosomes are affected by the gene change (transposition), indicating that it did not occur de novo . The variant has not been described earlier. However, the CHD7 variant c.2690G>C at the same position has been demonstrated to cause HH 18 . PolyPhen-2 19 predicts the amino acid exchange p.Arg897Gln as “benign” with a score of 0.053 (sensitivity: 0.93; specificity: 0.64) in the HumVar model.
MLPA analysis of the infertility associated genes ANOS1 , FGFR1 , PROK2 , PROKR2, CHD7 , GNRH1, GNRHR , and KISS1R excluded large duplications and deletions.
Discussion and conclusions
We identified the potentially disease causing familial heterozygous KISS1 variant c.-7C>T in two brothers diagnosed with HH. Since the mutation occurred in two brothers, we assume that it was inherited. The single nucleotide polymorphism (SNP) at position -7 is located in the 5’-UTR within the Kozak consensus sequence, which is a eukaryotic ribosome binding site in close proximity to the translational start codon 20 . Therefore, the mutation potentially interferes with proper ribosome binding and translation of the Kp protein. Kp deficiency is associated with disturbance of the hypothalamic-pituitary-gonadal axis which can result in HH 7 8 .
Interestingly, the KISS1 variant c.-7C>T has already been described in a patient with Kallmann syndrome 17 , which is a form of congenital HH that arises due to a lack of functional hypothalamic GnRH neurons and is characterized by deficiency of GnRH and sex steroid hormones as well as hypo- or anosmia 21 . Hypo- or anosmia occurs due to disturbance of the olfactory bulb development during embryogenesis. This results in aberrant migration of hypothalamic GnRH neurons, which originate in the medial olfactory placode 22 23 . A variety of gene mutations, including KISS1 mutations, are associated with the pathogenesis of Kallmann syndrome 17 24 25 26 . However, isolated Kp deficiency in patient 1 and 2 presumably does not affect embryonic olfactory bulb development and GnRH neuron migration, explaining why both patients have a normal sense of smell.
Since both patients presented with similar symptoms, it is not clear to what extent the CHD7 variant c.2690G>A identified in patient 2 contributed to the pathogenesis of HH. PolyPhen-2 classifies the mutation as benign. Nevertheless, this does not allow a prediction of the phenotype. The previously described CHD7 variant c.2690G>C affects the same position and has been shown to cause HH 18 . Therefore, the CHD7 mutation in patient 2 might have enhanced the pathogenesis of HH.
In conclusion, identification of the heterozygous KISS1 mutation c.-7C>T added another genetic variant to the list of HH causing genome aberrations 24 , that will facilitate to identify the cause of male idiopathic HH. The role of the CHD7 variant c.2690G>A in the pathogenesis of HH remains unclear.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Acevedo-Rodriguez A Kauffman A S Cherrington B D Emerging insights into hypothalamic-pituitary-gonadal axis regulation and interaction with stress signalling J Neuroendocrinol 201830 e 1259010.1111/jne.1259029524268 PMC 6129417 · doi ↗ · pubmed ↗
- 2Sykiotis G P Pitteloud N Seminara S B Deciphering genetic disease in the genomic era: the model of Gn RH deficiency Sci Transl Med 2010232 rv 210.1126/scitranslmed.3000288 PMC 393624820484732 · doi ↗ · pubmed ↗
- 3Boehm U Bouloux P-M Dattani M T Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment Nat Rev Endocrinol 20151154756410.1038/nrendo.2015.11226194704 · doi ↗ · pubmed ↗
- 4Sykiotis G P Plummer L Hughes V A Oligogenic basis of isolated gonadotropin-releasing hormone deficiency Proc Natl Acad Sci USA 2010107151401514410.1073/pnas.100962210720696889 PMC 2930591 · doi ↗ · pubmed ↗
- 5Vezzoli V Hrvat F Goggi G Genetic architecture of self-limited delayed puberty and congenital hypogonadotropic hypogonadism Front Endocrinol 2023131.069741 E 610.3389/fendo.2022.1069741 PMC 988469936726466 · doi ↗ · pubmed ↗
- 6Choe H K Kim H-D Park S H Synchronous activation of gonadotropin-releasing hormone gene transcription and secretion by pulsatile kisspeptin stimulation Proc Natl Acad Sci USA 20131105677568210.1073/pnas.1213594110023509283 PMC 3619287 · doi ↗ · pubmed ↗
- 7Seminara S B Messager S Chatzidaki E E The GPR 54 Gene as a Regulator of Puberty N Engl J Med 20033491614162710.1056/NEJ Moa 03532214573733 · doi ↗ · pubmed ↗
- 8De Roux N Genin E Carel J-C Hypogonadotropic hypogonadism due to loss of function of the Ki SS 1-derived peptide receptor GPR 54Proc Natl Acad Sci USA 2003100109721097610.1073/pnas.183439910012944565 PMC 196911 · doi ↗ · pubmed ↗