Novel loss-of-function SPAG17 homozygous variant segregated in a family with severe asthenozoospermia: upgrading gene-disease validity to strong
Li Wang, Jinli Li, Ling Huang, Jialing Wang, Li Zhou, Li Ding, Jia Li, Qinghua Zhang, Junyu Zhang, Guangmei Xie

TL;DR
A new genetic variant in SPAG17 was found to cause severe sperm motility issues in two brothers, confirming its role in male infertility.
Contribution
A novel homozygous SPAG17 variant was identified and validated as a strong contributor to severe asthenozoospermia.
Findings
A homozygous nonsense variant in SPAG17 (c.2188C>T; p.Q730*) was found in two siblings with severe asthenozoospermia.
Sperm analysis showed reduced motility, malformed flagella, and axonemal defects in affected individuals.
SPAG17's gene-disease validity for severe asthenozoospermia was upgraded to 'Strong' with a cumulative score of 13.4.
Abstract
Severe asthenozoospermia is a significant cause of male infertility, commonly associated with genetic defects affecting sperm motility. However, the specific genetic contributors remain underexplored. This study aimed to identify a genetic variant responsible for severe asthenozoospermia in two siblings and to evaluate the clinical validity of the gene-disease relationship between SPAG17 and this condition. Whole exome sequencing (WES) was performed on two siblings diagnosed with severe asthenozoospermia. Sperm motility and morphology were assessed through standard semen analysis and transmission electron microscopy (TEM). The gene-disease validity was evaluated using the ClinGen Gene–Disease Validity SOP, incorporating both genetic and experimental evidence. A novel homozygous nonsense variant in SPAG17 (NM_206996.4: c.2188C>T; p.Q730*) was identified in both affected siblings.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
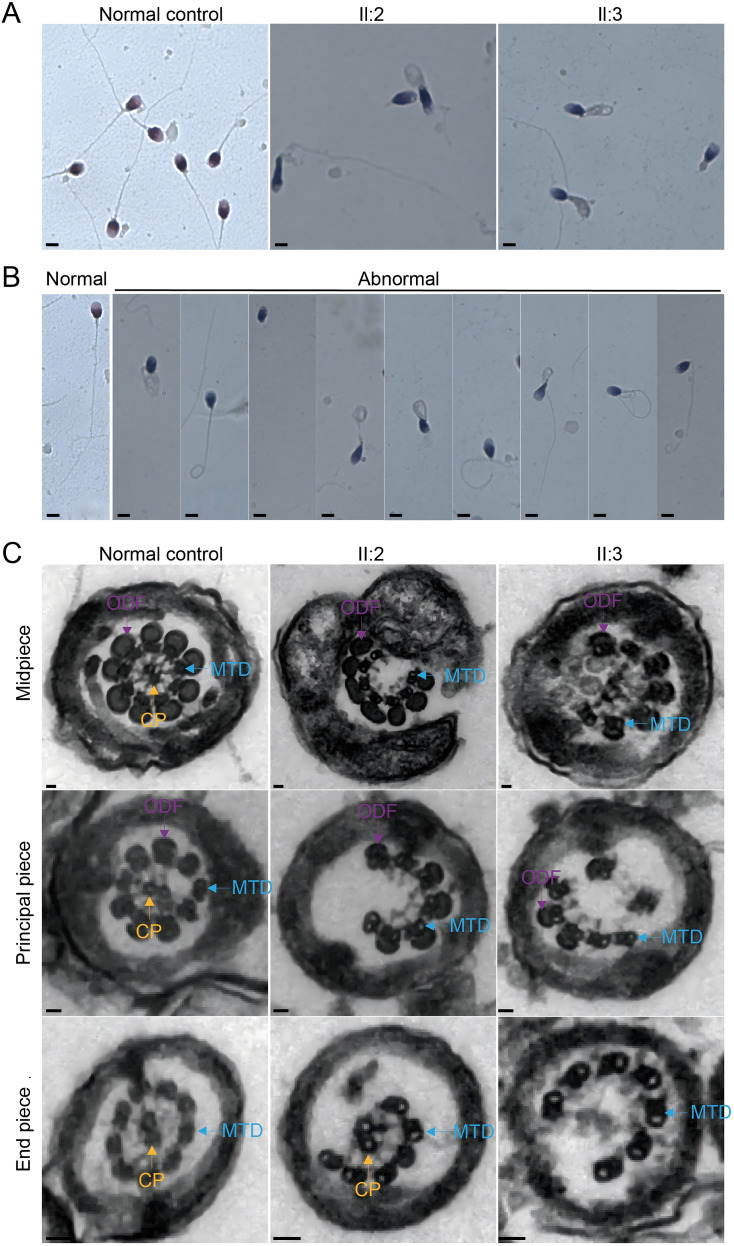 Figure 1
Figure 1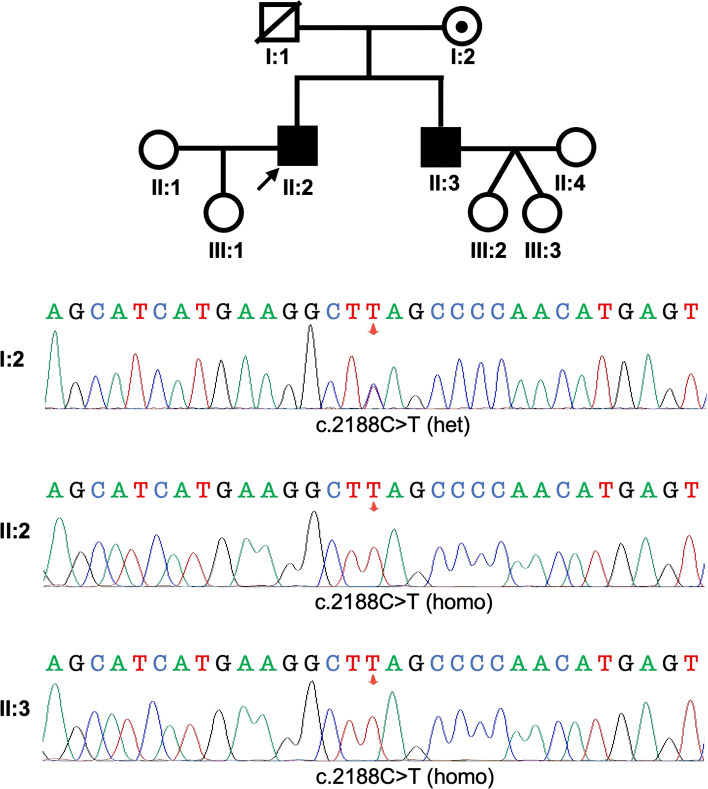 Figure 2
Figure 2| Information | II:2 | II:3 | Reference values |
|---|---|---|---|
| Genotype | MT/MT | MT/MT | |
| Age (years) | 37 | 34 | |
| Years of marriage | 10 | 3 | |
| BMI | 23.67 | 19.59 | |
| Semen parameters | |||
| Semen volume (ml) | 3.47±1.19 | 1.86±0.21 | >1.5 |
| Semen pH | Alkaline | Alkaline | Alkaline |
| Sperm concentration (106/ml, Mean ± SD) | 45.7±23.11 | 77.02±39.40 | >15 |
| Motile sperm (%, Mean ± SD) | 3.33±1.15 | 3.20±1.79 | >40 |
| Progressively motile sperm (%, Mean ± SD) | 2.33±0.58 | 0.8±0.45 | >32 |
| Sperm flagellar morphology | |||
| Absent flagella (%) | 22.79 | 10.07 | <5.0 |
| Short flagella (%) | 15.44 | 11.51 | <1.0 |
| Coiled flagella (%) | 23.53 | 22.3 | <17.0 |
| Angulation (%) | 16.91 | 20.14 | <13.0 |
| Irregular caliber (%) | 13.23 | 27.34 | <2.0 |
| Normal flagella (%) | 8.09 | 8.63 | >23.0 |
| Individual | Age at diagnosis | Variant in | Exon | Clinical diagnosis | Inheritance | Reference |
|---|---|---|---|---|---|---|
| II:5 | 29 | 30 | Severe asthenozoospermia | Homozygous | ( | |
| II:7 | 29 | 30 | Severe asthenozoospermia | Homozygous | ( | |
| (Patient 1)IV:1 | 30 | 6 | multiple morphological abnormalities of the flagella | Homozygous | ( | |
| (Patient 2)IV:3 | 25 | 6 | multiple morphological abnormalities of the flagella | Homozygous | ( | |
| (Patient 3)IV:1 | 42 | 15 | multiple morphological abnormalities of the flagella | Homozygous | ( | |
| (Patient 4)IV:4 | 33 | 15 | multiple morphological abnormalities of the flagella | Homozygous | ( | |
| Patient 1 | 35 | − | oligoasthenoteratozoospermia | Homozygous | ( | |
| Patient 1 | 34 | 15 | Severe asthenozoospermia | Homozygous | This study | |
| Patient 2 | 37 | 15 | Severe asthenozoospermia | Homozygous | This study |
| Genetic evidence: case-level data | |||||
|---|---|---|---|---|---|
| Evidence type | Case information | Suggested upgrades | Points given | References/Notes | |
| Functional data |
| ||||
| Variant Evidence: Autosomal Dominant*2 | Predicted or proven null variant (default 1.5 points, scoring range 0-3 points per variant) | +0.5 points | +0.5 points | 9.0 points | Two homozygous frameshift variants in |
| Other variant type with some evidence of gene impact (default 0.1 points, scoring range 0-1.5 points per variant) | +0.4 points | +0.4 points | 0.4 | Two homozygous missense variants in | |
| Segregation Evidence | Evidence of segregation in one or more families (scoring range 0-3 points) | 0 | No evidence available | ||
| Genetic Evidence: case-control data | |||||
| Case-Control Study Type | Case-Control Quality Criteria | Suggested Points/Study | Points Given | References/Notes | |
| Single Variant Analysis | •Variant detection methodology | 0-6 points | 0 | No evidence available | |
| Aggregate Variant Analysis | 0-6 points | 0 | No evidence available | ||
| Total genetic evidence points | 9.4 | ||||
| Experimental evidence | |||||
|---|---|---|---|---|---|
| Evidence category | Evidence type | Suggested points | Points given | References/Notes | |
| Default | Range | ||||
| Function | Biochemical function | 0.5 | 0-2 | 1.0 | |
| Protein interaction | 0.5 | 0-2 | 0 | No evidence available | |
| Expression | 0.5 | 0-2 | 0.5 | SPAG17 is highly expressed in testis tissue, where spermatogenesis takes place ( | |
| Functional Alteration | Patient cells | 1 | 0-2 | 0 | No evidence available |
| Non-patient cells | 0.5 | 0-1 | 0.5 | Primary cilia in chondrocytes, osteoblasts, and embryonic fibroblasts (MEFs) from knockout mice were shorter, and fewer cells had primary cilia compared to those from wild-type mice ( | |
| Models | Non-human model organism | 2 | 0-4 | 2 | Spag17 knockout mice are infertile due to a severe defect in spermatogenesis, with spermatogenesis arrested at the spermatid stage. The few sperm retrieved from the cauda epididymis were immotile and exhibited abnormal tail and head morphology ( |
| Cell culture model | 1 | 0-2 | 0 | No evidence available | |
| Rescue | Rescue in human | 2 | 0-4 | 0 | No evidence available |
| Rescue in non-human model organism | 2 | 0-4 | 0 | No evidence available | |
| Rescue in cell culture model | 1 | 0-2 | 0 | No evidence available | |
| Rescue in patient cells | 1 | 0-2 | 0 | No evidence available | |
| Total experimental evidence points | 4.0 | ||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSperm and Testicular Function · Hereditary Neurological Disorders · Genomics and Rare Diseases
Introduction
1
Sperm motility is a crucial determinant of fertility, which represents a fundamental feature of flagellate spermatozoa and playing a vital role in the fertilization process (1). Asthenozoospermia is a major cause of male infertility, affecting approximately 19% of infertile patients. It is defined by a progressive motility (PR) rate of spermatozoa below 32% (2). Asthenozoospermia has been attributed to various factors, including genetic causes, lifestyle, environmental pollutants, prolonged sexual abstinence, partial blockage of the seminal tract, varicocele, and infections (3–6). Although genetic deficiencies are significant causal factors (7), the specific genetic contributors to this condition remain largely unexplored.
SPAG17 encodes a protein located in the axoneme central pair complex of motile cilia and flagella (8). Previous studies in chlamydomonas have demonstrated that PF6, the ortholog of the mammalian SPAG17 protein, is critical for flagellar motility (9). Spag17 knockout mice exhibit infertility due to a severe defect in spermatogenesis. Histological analysis of testis sections from mutant mice revealed seminiferous tubules with spermatogenesis arrested at the spermatid stage. The few sperm retrieved from the cauda epididymis were immotile and exhibited abnormalities morphology (8). According to reports, several male cases with infertility carry homozygous variants in the SPAG17 gene (10–12). These findings collectively underscore the critical role of SPAG17 in sperm development, motility, and male fertility.
Despite this accumulating evidence, a formal evaluation of the clinical validity of the gene-disease association between SPAG17 and severe asthenozoospermia has not been thoroughly performed. Evaluating the clinical validity of the gene-disease relationship is essential for variant classification and clinical interpretation (13). The Clinical Genome Resource (ClinGen) has developed a semiquantitative points-based framework to assign clinical validity classifications to gene–disease relationships (14, 15). Clinical validity is categorized as definitive (12–18 points, replicated over time), strong (12–18 points), moderate (7–11 points), limited (0.1–6 points), no known disease relationship (0 points), disputed (contradictory evidence), or refuted (contradictory evidence outweighs supportive evidence). The ACMG recommends that variants in moderate genes should generally not be classified higher than Likely Pathogenic, and variants in genes with poorly understood disease-gene relationships (Limited, Disputed, Refuted) should not be classified higher than variants of uncertain significance (16).
In this study, we identified a novel homozygous nonsense variant in SPAG17 in two siblings with severe asthenozoospermia significantly contributes to understanding the genetic basis of this condition. The detailed semen analysis and ultrastructural studies revealed profound abnormalities in sperm morphology and flagellar structure, further supporting the involvement of SPAG17 in male infertility. This study, in conjunction with previous reports and emerging reports, substantially enhances the evidence for the gene-disease relationship between SPAG17 and severe asthenozoospermia, elevating the association to a strong strength according to the ClinGen Gene–Disease Validity SOP. These findings underscore the importance of SPAG17 in spermatogenesis and male fertility, providing valuable insights into potential diagnostic and therapeutic approaches for affected individuals.
Methods
2
Patients and ethics statement
2.1
This study received ethical approval from the Gansu Provincial Maternal and Child Health Hospital (Gansu Provincial Central Hospital), and informed consent was obtained from each participant. The brothers with primary infertility and their mother were recruited from the Gansu Provincial Maternal and Child Health Hospital (Gansu Provincial Central Hospital).
Whole exome sequencing, variant calling, and sequence variant validation
2.2
Genomic DNA was isolated from peripheral blood lymphocytes of participants using QIAamp DNA Blood Kits (Qiagen, Hilden, Germany). Library preparation and exome enrichment were conducted using SureSelect Human All Exon V6 (Agilent Technologies, Santa Clara, CA, USA). The prepared libraries were sequenced using the Illumina NovaSeq^®^ 6000 system (Illumina, San Diego, CA, USA). The variants were filtered and prioritized using TGex (https://fa.shanyint.com/, accessed 4 January 2025) (17). The gene variants detected by WES were validated by Sanger sequencing, using the following primers for amplification and sequencing: SPAG17-c2188-F=CCGAGAACCTTCAGATCCTAGTC and SPAG17-c2188-R=AGGGTTGAAATAAAGAGAACTTATGG.
Literature review and gene–disease clinical validity curation
2.3
A comprehensive literature search was conducted using PubMed and Google Scholar to identify publications on the relationship between SPAG17 and male infertility in humans and animal models. Only English-language manuscripts and abstracts were considered. Cases reporting deleterious SPAG17 variants associated with male infertility due to asthenozoospermia were reviewed. The strength of the gene–disease association between SPAG17 and autosomal recessive severe asthenozoospermia was curated following the ClinGen Gene–Disease Validity SOP, version 11 (18).
Semen parameters and sperm morphology analysis
2.4
Semen samples were collected by masturbation and incubated at 37 °C for 30 minutes to achieve liquefaction. Routine semen analysis (volume, sperm concentration, and motility) was performed using the Sperm Class Analyzer CASA System (SCA, Spain) following World Health Organization guidelines (5th Edition) (19). Semen samples from subjects were washed with PBS, fixed in 4% paraformaldehyde (PFA), and stained with Mayer’s hematoxylin and a 1% eosin Y solution (FUJIFILM WakoPure Chemical). Sperm morphology was assessed by examining at least 130 sperm from each sample.
Transmission electron microscopy
2.5
Spermatozoa were immersed in a 2.5% glutaraldehyde solution, washed three times with 0.1 mol/L phosphate buffer (PB, pH 7.2), and postfixed with 1% osmium tetroxide in 0.1 mol/L PB for 1–1.5 hours at 4 °C. Dehydration was performed sequentially using ethanol solutions at concentrations of 50%, 70%, 80%, 95%, and 100%, followed by 100% acetone. Infiltration was then carried out by immersing the samples overnight at 37 °C in a 1:1 mixture of acetone and SPI-Chem resin (containing dodecenyl succinic anhydride, N-methylacetamide, SPI-Pon 812, and DMP-30). After infiltration and embedding in Epon 812 resin, ultrathin sections were stained with uranyl acetate and lead citrate. These sections were then examined and photographed using a TEM (TECNAI-10, Philips) operating at an accelerating voltage of 80 kV.
Normal controls
2.6
For comparative analyses in semen morphology and ultrastructural studies, normal control samples were obtained from three healthy fertile volunteers with normal semen parameters (progressive motility > 40%, normal morphology > 4%) and no history of reproductive disorders. Control samples were processed identically and simultaneously with patient samples under the same laboratory conditions, including fixation, staining, and TEM preparation, to minimize batch effects and pre-analytical variability.
Result
3
Severe asthenozoospermia and ultrastructural abnormalities in infertile two siblings
3.1
The patient and his sibling, with primary infertility durations of 10 and 3 years, respectively, both exhibit severe asthenozoospermia. Sperm motility and progressive motility of them were both significantly below the normal reference values (Table 1). Hematoxylin and eosin (H&E) staining was utilized to evaluate sperm morphology. The sperm from the two patients both exhibited a reduced proportion of morphologically normal sperm and an increased prevalence of abnormalities in flagella, including absent, short, curly, angular, and irregularly calibrated flagella (Figure 1, Table 1). Additionally, transmission electron microscopy (TEM) was used to analyze the ultrastructure of spermatozoa from the two siblings. Strikingly, compared to the regular “9 + 2” axonemal arrangement observed in sperm flagella from normal controls, the spermatozoa from the two patients exhibited absent or disorganized central-pair microtubules (CPs) and irregular or absent outer dense fibers (ODFs) and microtubule doublets (MTDs) in the flagellar midpiece. Additionally, the principal piece showed a loss of most axonemal microtubules, which were irregularly arranged, and the peripheral MTDs or CPs were absent in the end piece.
Morphological and ultrastructural analysis of spermatozoa in normal controls and individuals with bi-allelic SPAG17 variants. (A) Morphology of the spermatozoa from normal control and men harboring bi-allelic SPAG17 variants (scale bars, 15 μm). (B) Papanicolaou staining revealed sperm with normal morphology and various abnormal morphologies (scale bars, 15 μm). (C) Representative TEM micrographs display cross-sections of the midpiece, principal piece, and end piece of sperm flagella from normal controls and individuals with bi-allelic SPAG17 variants. The axoneme structure, along with its accessory components illustrated in the diagram, includes peripheral microtubule doublets (DMT), central pairs (CP), and outer dense fibers (ODF) (scale bars, 150 nm).
Additionally, transmission electron microscopy (TEM) was used to analyze the ultrastructure of spermatozoa from the two siblings. A total of 60 cross-sectional flagellar images per individual were evaluated. Compared to the regular “9 + 2” axonemal arrangement observed in controls, patient spermatozoa exhibited absent central-pair microtubules in 82% of cross-sections, disorganized outer dense fibers in 75%, and irregular microtubule doublets in 68%. Representative images are shown in Figure 1C.
Identification of a homozygous loss-of-function variant in SPAG17 in two siblings affected by severe asthenozoospermia.
3.2
Prior to referral, both individuals had underwent unsuccessful intracytoplasmic sperm injection (ICSI) cycles at an external clinic. Subsequent diagnostic genetic testing identified an identical homozygous nonsense variant in SPAG17 (NM_206996.4: c.2188C>T; p.Q730*) in both siblings, resulting in a premature stop codon. Sanger sequencing confirmed homozygosity for the SPAG17 variant in both patients and revealed that their mother was heterozygous for the same variant (Figure 2), consistent with autosomal recessive inheritance. The father, now deceased, could not be tested due to the unavailability of DNA. Following another ICSI cycle combined with assisted oocyte activation (AOA), both individuals achieved live births: II:2 had a daughter, and II:3 had twin daughters.
Identification of a homozygous SPAG17 variant in two infertile siblings with severe asthenozoospermia. Top: Pedigree of a family with two infertile males (II:2 and II:3), highlighting the proband indicated by an arrow. Bottom: Sanger sequencing chromatograms are shown for the tested individuals. The affected males harbor a homozygous loss-of-function variant in SPAG17.
The addition of this case upgrades the gene–disease validity between SPAG17 and autosomal recessive severe asthenozoospermia from “Moderate” to “Strong”
3.3
The identification of a novel homozygous loss-of-function variant in SPAG17 associated with severe asthenozoospermia prompted a formal evaluation of the gene-disease validity between SPAG17 and severe asthenozoospermia. The curation considered both genetic and experimental evidence. The current cases contributed significant genetic evidence, with 3 points awarded based on the identification of a predicted null variant in an autosomal recessive condition. This was combined with evidence from 4 previous report male cases with infertility harboring two homozygous missense variants and two homozygous loss of function variants in SPAG17 (Table 2) (10–12). Collectively, the cumulative genetic evidence scored 9.4 out of a total of 12 points (Table 3) (15, 18).
The correlation between SPAG17 and severe asthenozoospermia is also supported by experimental evidence (Table 4). SPAG17 is crucial for the structure and function of motile cilia (20) and facilitates the translocation of protamines from the cytoplasm to the nucleus, a key process in sperm production (21). SPAG17 is highly expressed in testicular tissue, where spermatogenesis occurs (22). In SPAG17 knockout mice, primary cilia in chondrocytes, osteoblasts, and embryonic fibroblasts (MEFs) were shorter, and fewer cells exhibited primary cilia compared to wild-type mice (23). Spag17 knockout mice are infertile due to a severe spermatogenesis defect, with spermatogenesis arrested at the spermatid stage. Sperm retrieved from the cauda epididymis were immotile and displayed abnormal tail and head morphology (8). These data underscore the critical role of SPAG17 in male reproductive biology, with a cumulative evidence score of 4 out of 6 allowable points according to the ClinGen Gene–Disease Validity SOP (Table 4) (13, 18).
Combining the genetic (9.4 points) and experimental (4 points) evidence resulted in a total score of 13.4 points. According to the ClinGen Gene–Disease Validity SOP, the 3 points contributed by our cases upgraded the gene–disease validity between SPAG17 and autosomal recessive severe asthenozoospermia from “Moderate” (requiring 7–11 points) to “Strong” (requiring 12–18 points).
Discussion
4
The identification of a novel homozygous nonsense variant in SPAG17 (NM_206996.4:c.2188C>T; p.Q730*) in these two siblings with severe asthenozoospermia significantly strengthens the genetic evidence implicating SPAG17 in this specific form of male infertility. Our finding, along with the previously reported homozygous missense variant (p. R1448Q) in the SPAG17 gene in twins with severe asthenozoospermia (11), and the more recently identified homozygous variants (c.4511A>G, p.Asn1504Ser; c.829 + 1G>T, p.Asp212_Glu276del; c.2120del, p.Leu707*) associated with oligoasthenoteratozoospermia (OAT) and MMAF phenotypes (10, 12), provides genetic evidence of SPAG17’s crucial role in sperm motility and male infertility. The sperm motility, morphology, and ultrastructure observed in the patients analyzed in this study align closely with findings from prior animal model research on SPAG17. Sperm motility analysis revealed severely reduced progressive motility, consistent with the immotile sperm phenotype reported in Spag17 knockout mice (8). Morphological evaluation demonstrated abnormalities, such as absent, short, and irregularly shaped flagella, consistent with defective sperm tails in knockout mice (8). Additionally, ultrastructural examination demonstrated disruptions in the “9 + 2” axonemal arrangement, including disorganized or absent central-pair microtubules, outer dense fibers, and microtubule doublets, consistent with flagellar defects reported in Spag17 knockout animal models (8). In addition to animal knockout models, other experimental evidence further supports the link between SPAG17 and severe asthenozoospermia. SPAG17 is essential for the structure and function of motile cilia and for the translocation of protamines during sperm production (20, 21). It is highly expressed in testicular tissue, where spermatogenesis occurs (22). The consistent findings of significantly reduced sperm motility and the array of flagellar abnormalities, both at the microscopic and ultrastructural levels, directly mirror the phenotypes observed in Spag17 knockout animal models, further reinforcing the functional consequence of SPAG17 deficiency in humans.
The central significance of our study lies in its impact on the formal evaluation of the gene-disease validity between SPAG17 and severe asthenozoospermia. Prior to this study, while evidence from Spag17 knockout mouse models and human case report (8, 10–12) suggested a link, the overall gene-disease validity, as assessed by the rigorous ClinGen Gene-Disease Validity SOP, was classified as “Moderate”(10.4 points). Our identification of this novel, loss-of-function homozygous variant in two affected siblings provided the critical additional genetic evidence needed to strengthen the association. Our cases, contributing a significant 3 points to the genetic evidence score within the ClinGen framework, effectively upgraded the overall evidence beyond the threshold required for “Strong”(13.4 points).
This upgrade from “Moderate” to “Strong” has significant implications for the clinical management of male infertility. Firstly, it provides more substantial support for including SPAG17 in diagnostic gene panels for asthenozoospermia (16), enhancing the precision of genetic testing and improving the likelihood of identifying the underlying genetic cause in similar cases. Secondly, the “Strong” classification has a direct impact on SPAG17 variant in clinical settings. Variants in genes with “Moderate” validity could be classified as “Likely Pathogenic”, in contrast, for genes with a disease association categorized as definitive or strong, variants can be classified as high as pathogenic (16). This shift provides more actionable information for patient management and reproductive planning.
Notably, the clinical management of the affected siblings in our study provides further insight into the multifaceted role of SPAG17. Both individuals underwent ICSI combined with assisted oocyte activation (AOA), suggesting potential fertilization impairment beyond mere motility defects. SPAG17 is known to facilitate nuclear translocation of protamines during spermiogenesis, and its deficiency may lead to chromatin abnormalities and impaired sperm-derived oocyte activation capacity. This aligns with emerging evidence that flagellar structural proteins can also influence sperm nuclear integrity and centriolar function. Therefore, AOA may address not only the overt motility barrier but also underlying chromatin or activation deficiencies in SPAG17-deficient sperm.
In summary, we identified a novel homozygous loss-of-function variant in SPAG17 in two affected siblings. Semen analysis and ultrastructural studies have revealed a significant reduction in sperm motility and severe morphological abnormalities in affected patients. Combined with previous genetic and experimental evidence, these findings upgrade the gene-disease validity for SPAG17-related autosomal recessive severe asthenozoospermia to “Strong”. Our findings highlight the critical role of SPAG17 in male fertility and have significant clinical implications, supporting the inclusion of SPAG17 in male infertility screening gene panels, enabling more reliable variant classification, and ultimately enabling more precise genetic counseling and therapeutic strategies for affected individuals.
Study limitations and future perspectives
4.1
This study has certain limitations. Most notably, due to the scarcity and complete consumption of patient sperm samples for essential diagnostic and ultrastructural analyses (e.g., TEM), we were unable to perform direct protein-level validation, such as immunofluorescence or Western blotting, to confirm the absence of the SPAG17 protein. While the identification of a homozygous nonsense variant provides strong genetic evidence for a loss-of-function mechanism, future studies with access to relevant samples are encouraged to validate these findings at the protein level. Additionally, the functional link between SPAG17 deficiency and the observed need for assisted oocyte activation warrants further mechanistic investigation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Holt WV Van Look KJW . Concepts in sperm heterogeneity, sperm selection and sperm competition as biological foundations for laboratory tests of semen quality. Reproduction. (2004) 127:527–35. doi: 10.1530/rep.1.00134, PMID: 15129008 · doi ↗ · pubmed ↗
- 2Zheng J Lu Y Qu X Wang P Zhao L Gao M . Decreased sperm motility retarded ICSI fertilization rate in severe oligozoospermia but good-quality embryo transfer had achieved the prospective clinical outcomes. P Lo S One. (2016) 11:e 0163524. doi: 10.1371/journal.pone.0163524, PMID: 27661081 PMC 5035010 · doi ↗ · pubmed ↗
- 3Salas-Huetos A BullóM Salas-SalvadóJ . Dietary patterns, foods and nutrients in male fertility parameters and fecundability: A systematic review of observational studies. Hum Reprod Update. (2017) 23:371–89. doi: 10.1093/humupd/dmx 006, PMID: 28333357 · doi ↗ · pubmed ↗
- 4Tournaye H Krausz C Oates RD . Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol. (2017) 5:544–53. doi: 10.1016/S 2213-8587(16)30040-7, PMID: 27395771 · doi ↗ · pubmed ↗
- 5Adams JA Galloway TS Mondal D Esteves SC Mathews F . Effect of mobile telephones on sperm quality: A systematic review and meta-analysis. Environ Int. (2014) 70:106–12. doi: 10.1016/j.envint.2014.04.015, PMID: 24927498 · doi ↗ · pubmed ↗
- 6Ortega C Verheyen G Raick D Camus M Devroey P Tournaye H . Absolute asthenozoospermia and ICSI: What are the options? Hum Reprod Update. (2011) 17:684–92. doi: 10.1093/humupd/dmr 018, PMID: 21816768 · doi ↗ · pubmed ↗
- 7Sudhakar DVS Shah R Gajbhiye RK . Genetics of male infertility - present and future: A narrative review. J Hum Reprod Sci. (2021) 14:217–27. doi: 10.4103/jhrs.jhrs_115_21, PMID: 34759610 PMC 8527069 · doi ↗ · pubmed ↗
- 8Kazarian E Son H Sapao P Li W Zhang Z Strauss JF . SPAG 17 is required for male germ cell differentiation and fertility. Int J Mol Sci. (2018) 19:1252. doi: 10.3390/ijms 19041252, PMID: 29690537 PMC 5979577 · doi ↗ · pubmed ↗