Comparative Genomics and Phylogenomics of the Mustelinae Lineage (Mustelidae, Carnivora)
Azamat A Totikov, Andrey A Tomarovsky, Polina L Perelman, Tatiana M Bulyonkova, Natalia A Serdyukova, Aliya R Yakupova, David Mohr, Daniel W Foerster, Jose Horacio Grau Jipoulou, Violetta R Beklemisheva, Mikhail Sidorov, Inês Miranda, Liliana Farelo, Alexei V Abramov

TL;DR
This study explores the evolutionary history and genetic diversity of the Mustelinae subfamily using whole-genome data from 10 species, revealing insights into their chromosomal evolution and demographic patterns.
Contribution
The paper provides the first comprehensive comparative and phylogenomic analysis of Mustelinae, including novel genome assemblies and insights into chromosomal evolution.
Findings
Marked homozygosity was observed in some Asian Mustelinae lineages, while widespread species showed high genetic diversity.
Phylogenomic analysis confirmed the split of M. richardsonii from M. erminea but found no speciation within M. nivalis.
Ancestral karyotypes for Mustela (2n = 44) and Mustelinae (2n = 42) were confirmed through chromosomal rearrangement analysis.
Abstract
Mustelinae are among the most diverse and taxonomically complex subfamilies within the Mustelidae, yet their evolutionary history and genetic diversity remain largely unexplored at the whole-genome level. Here, we present the first comprehensive comparative and phylogenomic study of this lineage, integrating nuclear and mitochondrial genomes from 10 species across the Holarctic and Indomalayan realms. Our dataset includes two novel genome assemblies (Mustela strigidorsa, M. sibirica) and an improved genome for M. nivalis, enabling robust cross-species analyses of genome size, chromosomal evolution, genetic diversity, and demographic history. We uncover striking inter- and intraspecific variation in genome-wide heterozygosity and genome size, with evidence of marked homozygosity in some Asian lineages (M. eversmanii, M. sibirica, M. strigidorsa) and remarkable genetic diversity in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Fig. 1
Fig. 1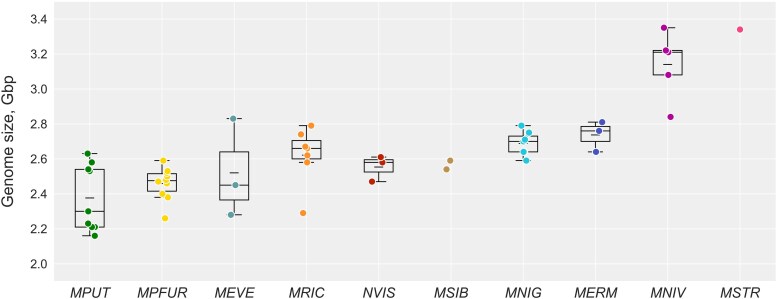 Fig. 2
Fig. 2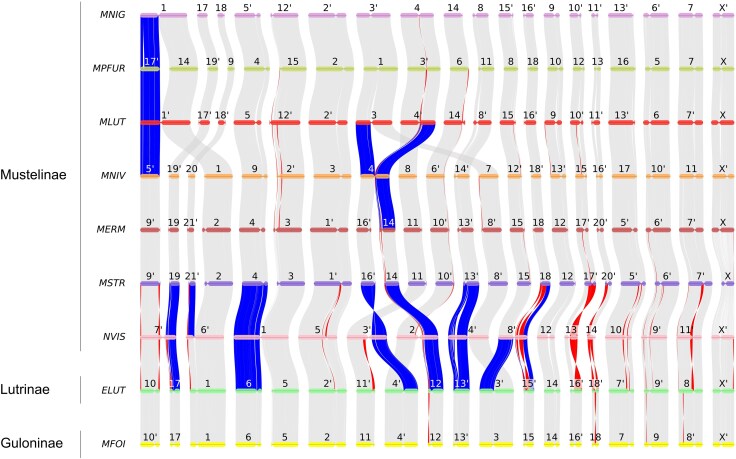 Fig. 3
Fig. 3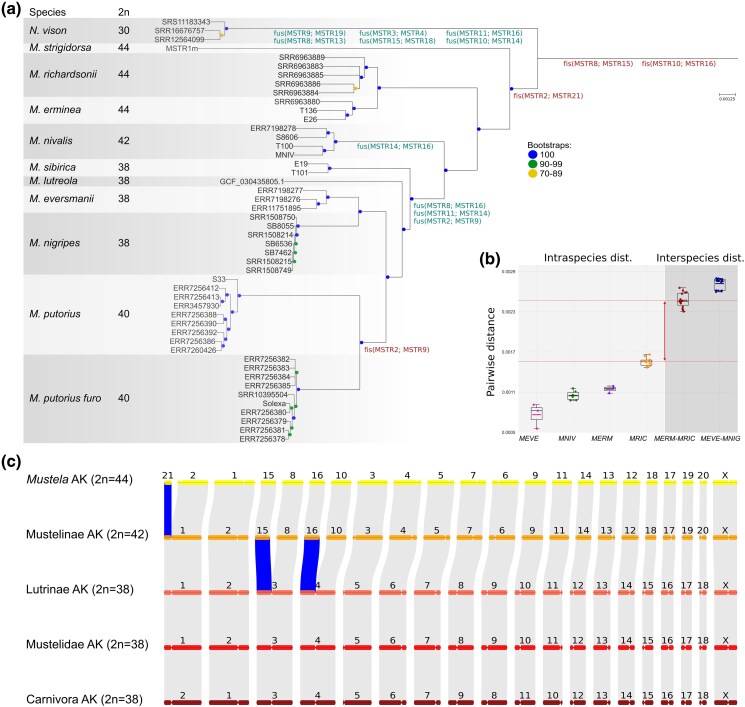 Fig. 4
Fig. 4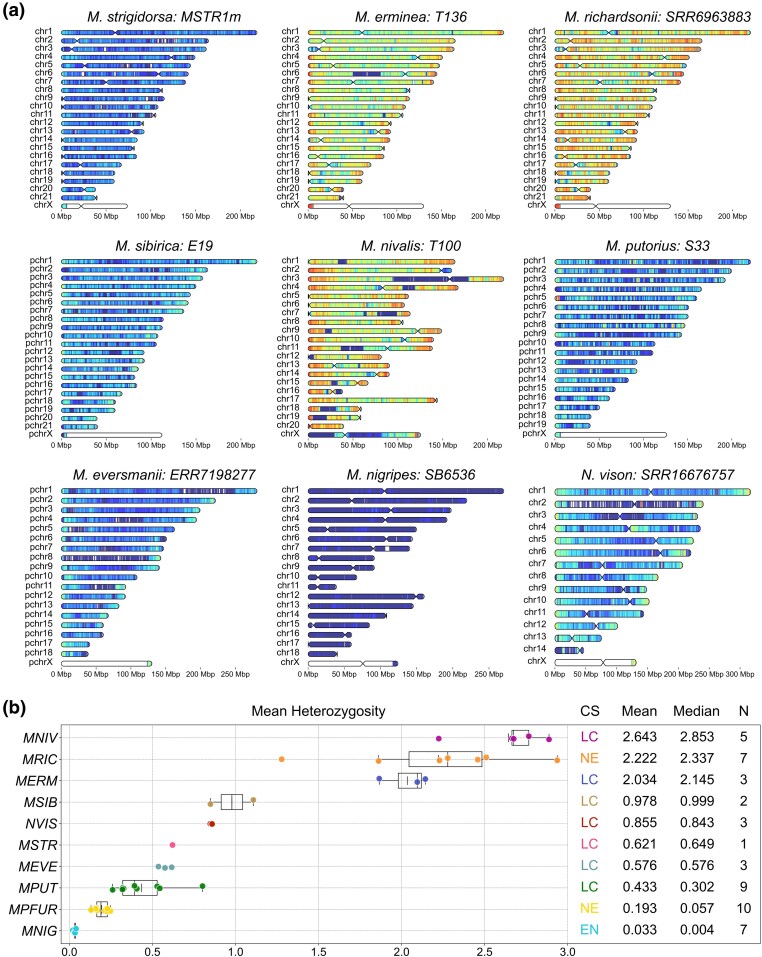 Fig. 5
Fig. 5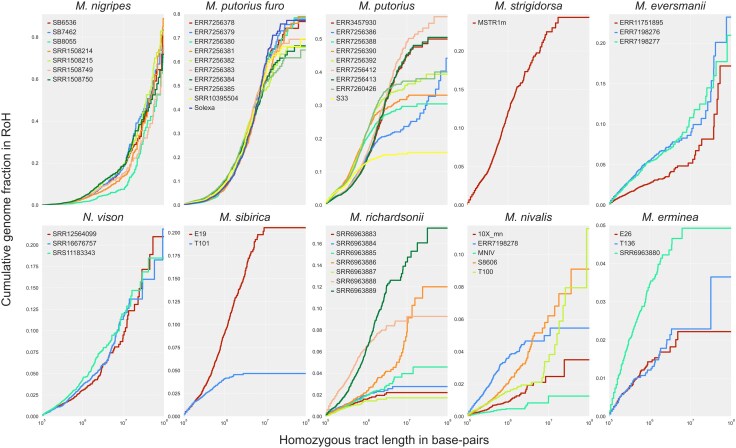 Fig. 6
Fig. 6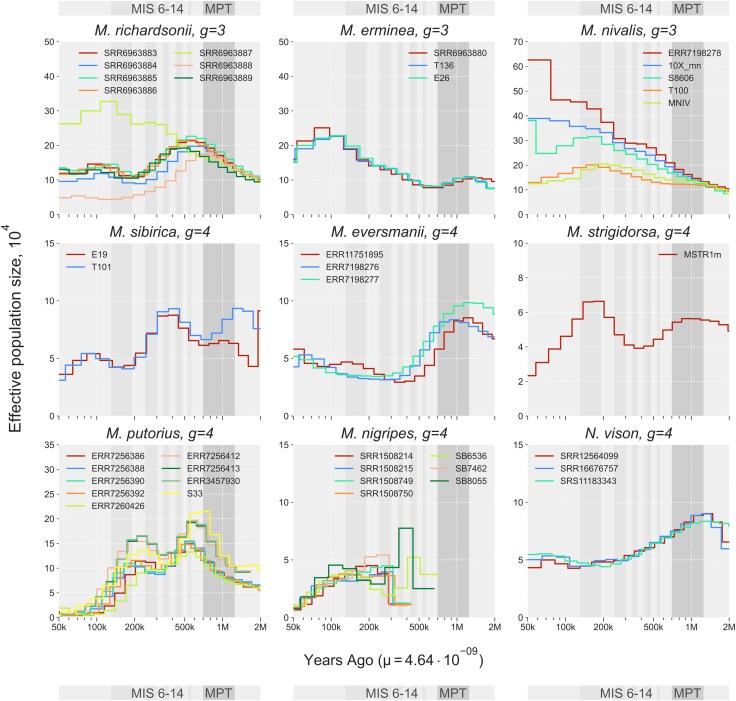 Fig. 7
Fig. 7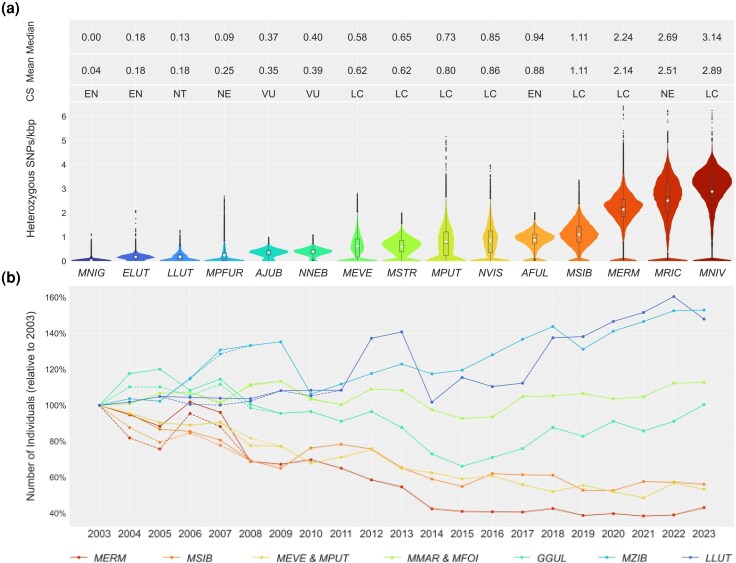 Fig. 8
Fig. 8- —Carlsbergfondet Research Infrastructure
- —Danish National Research Foundation10.13039/501100001732
- —ZIN10.13039/501100010763
- —Portuguese Fundação para a Ciência e a Tecnologia (FCT)
- —ERC-Portugal programme
- —FCT10.13039/100006129
- —Portuguese MCTES/FCT and ESF
- —St. Petersburg State University10.13039/501100004285
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEvolution and Paleontology Studies · Chromosomal and Genetic Variations · Genomics and Phylogenetic Studies
Introduction
The Mustelinae lineage represents the most numerous and diverse group of small mammals within the Mustelidae family, comprising at least 20 recognized species in the genera Mustela and Neogale (Fig. 1). Their high ecological adaptability has enabled them to inhabit a wide range of environments throughout the Holarctic region (Fig. 1) (Macdonald et al. 2017). While most Mustelinae species inhabit the Palearctic and Nearctic regions, some are restricted to the Neotropics (N. africana, N. felipei) or the Indomalayan zone (M. kathiah, M. strigidorsa, M. nudipes, and M. lutreolina). Many of these species are exploited by humans to varying degrees, whether in the fur industry (M. sibirica, M. putorius, M. eversmanii, M. erminea, and N. vison), in agriculture for rodent population control (e.g. M. erminea), in traditional Asian medicine (e.g. M. strigidorsa), as domestic pets, or even as model organisms for studying various viral diseases (M. putorius furo) (Belser et al. 2011; Harrington et al. 2017; Hiller and Vantassel 2022).
Geographic range and global conservation status of Mustelinae species. a–l) Photos of 12 out of the 20 currently recognized Mustelinae species reproduced from Flickr and iNaturalist under Creative Commons licenses: a) Least weasel (M. nivalis), Yersinia pestis, Flickr, CC BY-NC-SA; b) Stoat (M. erminea), Jing-Yi Lu, iNaturalist, CC BY-NC; c) American ermine (M. richardsonii), Vinny Pellegrino, iNaturalist, CC BY-NC; d) Back-striped weasel (M. strigidorsa), lordworm_cryptopsy, iNaturalist, CC BY-NC; e) Mountain weasel (M. altaica), Li Jianong, iNaturalist, CC BY-NC; f) Black-footed ferret (M. nigripes), Tory Mathis, iNaturalist, CC BY-NC; g) Steppe polecat (M. eversmanii), Anna Golubeva, iNaturalist, CC BY-NC-ND, reproduced with the permission of the author; h) Siberian weasel (M. sibirica), Nazim Ali Khan, iNaturalist, CC BY-NC; i) Yellow-bellied weasel (M. kathiah), jbhatia, iNaturalist, CC BY-NC; j) Long-tailed weasel (N. frenata), Donna Pomeroy, iNaturalist, CC BY-NC; k) European polecat (M. putorius), bandwidthbob, iNaturalist, CC BY-NC; l) American mink (N. vison), Tobin Brown, iNaturalist, CC BY-NC. Images were cropped for clarity. m) Geographic range and global conservation status of Mustelinae species. Ranges for M. richardsonii, M. haidarum, and M. aistoodonnivalis are based on previous studies (Colella et al. 2018, 2021; Liu et al. 2023). Ranges for all other species were obtained from the IUCN Red List of Threatened Species (IUCN 2025). Global conservation statuses according to IUCN data: LC, Least Concern, NT, Near Threatened, VU, Vulnerable, EN, Endangered, CR, Critically Endangered, NE, Not Evaluated.
Research on Mustelinae species has often focused only on describing their morphology, emphasizing physical characteristics such as body size, skull shape, fur patterns, and other distinctive features (Kitchener et al. 2017). However, these studies revealed the challenges of studying this subfamily, with factors such as varying degrees of sexual dimorphism, age-related variability, and the occurrence of interspecific hybrids complicating accurate taxonomic delineation (Kitchener et al. 2017). As a result, researchers increasingly turned to molecular and cytogenetic approaches to gain deeper insight into the evolution of the subfamily. Cytogenetic research elucidated the complex karyotypic evolution in Mustelinae, characterized by multiple chromosomal rearrangements, including fusions that produced the N. vison karyotype (2n = 30) and both fissions and fusions that generated a karyotype resembling that of M. erminea (2n = 44) (Graphodatsky et al. 1989). Moreover, substantial differences in the number of chromosomal arms among Mustela species, despite similar or identical diploid numbers (2n), cannot be explained solely by typical structural rearrangements (Graphodatsky et al. 1989). Karyotype evolution within Mustela has involved the emergence of additional, entirely heterochromatic arms in the karyotypes of certain species, through an increase in heterochromatic material on the short arms of originally acrocentric chromosomes (Graphodatsky et al. 2002). While Mustelinae species share some similarities in heterochromatin localization, they differ in the size of heterochromatic blocks and, consequently, in total heterochromatin content, which affects the genome size (Graphodatsky et al. 1977). These and other cytogenetic studies have laid the groundwork for understanding the complex phylogenetic relationships within this diverse group of mammals. Notably, even before the advent of sequencing technologies, cytogenetic studies had already indicated the need to separate Neogale from Mustela within the Mustelinae subfamily (Graphodatsky et al. 1989).
In recent decades, the systematics of Mustelidae species has undergone significant revisions and changes, primarily based on nuclear and/or mitochondrial data (Koepfli et al. 2008a; Yu et al. 2011; Sato et al. 2012; Law et al. 2018; Hassanin et al. 2021). Stepping beyond morphological markers has enabled a reevaluation of the traditional taxonomy of Mustelidae (Simpson 1945), leading to the division of the five previously recognized subfamilies into eight (Koepfli et al. 2008a; Yu et al. 2011). Subsequent studies focusing on Mustelinae, along with extensive discussions (Harding and Smith 2009; Hassanin et al. 2021; Patterson et al. 2021, 2025), helped to refine the boundaries between the genera Mustela and Neogale. Species-level classification remains challenging due to the rapid evolutionary diversification within this group (Koepfli et al. 2008b; Law et al. 2018; Hassanin et al. 2021), which has resulted in a number of morphologically indistinguishable species, as well as hard-to-resolve nodes on phylogenetic trees (Koepfli et al. 2008a). This complexity is further exacerbated by hybridization among some species (Cabria et al. 2011; Cserkész et al. 2021), which often leads to ambiguous phylogenetic relationships (Etherington et al. 2022). Uncertainty over species boundaries contributes to the lack of consensus on the total number of species or subspecies within Mustelinae. Despite these difficulties, a notable taxonomic revision has been the recognition of M. itatsi as taxonomically distinct from the closely related M. sibirica, previously classified as a single species (Abramov 2000). Similarly, taxonomic reassessment has successfully resolved the classification of M. erminea. Whole-genome and mitochondrial data (Colella et al. 2018, 2021) revealed that North American M. erminea actually comprises three distinct species: M. erminea, M. richardsonii, and M. haidarum. M. nivalis, the least weasel, has a similar range as ermines, but is still considered to be a single species. Multiple studies report a notable variation of phenotypic traits, eg, of body length and mass, coloration, and craniological metrics, but researchers do not agree on the number of subspecies (McDevitt et al. 2013; Vass and Bende 2023). Moreover, a recent phylogeographical study based on four mitochondrial and nuclear markers showed presence of two major clades correlated with the coat coloration, but have not reliably resolved low-level structure (Sato et al. 2020). These facts make a hypothesis of a species complex within M. nivalis deserving consideration and evaluation on the genomic level.
Currently, mitochondrial genome assemblies are available for nearly every species within the Mustelinae, with the exception of M. lutreolina, and M. aistoodonnivalis. However, the availability of whole-genome data is far less complete. Genome assemblies are absent for thirteen species (M. strigidorsa, M. sibirica, M. altaica, M. aistoodonnivalis, M. itatsi, M. kathiah, M. lutreolina, M. nudipes, M. haidarum, M. richardsonii, N. frenata, N. felipei, and N. africana). Among the remaining species, chromosome-level genome assemblies were published for only six species (M. nivalis, M. erminea, M. lutreola, M. nigripes, M. putorius furo, and N. vison), and scaffold-level genome assemblies exist for only two others (M. eversmanii and M. putorius). Notably, the genome assembly for M. nivalis was published very recently (O’Brien et al. 2025). Despite the increasing volume of available genomic data, its distribution across the Mustelinae lineage remains uneven (Table S1), underscoring the need for further sequencing and genome assembly efforts for the less-studied species.
Comparative analysis based on the integration of nuclear and mitochondrial genome data represents a powerful tool for studying evolutionary processes, enabling the identification of both common features and key differences between related species. This approach is particularly significant in the context of conservation research, where understanding genetic diversity, phylogenetic relationships, and demographic history is crucial for developing effective biodiversity conservation strategies. In this study, we compiled an extensive dataset, comprising 10 genome assemblies, 50 resequencing samples, and 149 mitochondrial genomes. This dataset includes three new genomic assemblies, two of which were generated by us and one improved from a publicly available draft genome assembly, as well as 9 samples newly resequenced as part of this project. Using this comprehensive dataset, we aimed to: (i) evaluate genome size variation within and between species; (ii) determine interspecific and intraspecific phylogenetic relationships; (iii) reconstruct synteny blocks to identify structural rearrangements among genomes; (iv) assess and compare genetic diversity among the species on the whole-genome level; and (v) reconstruct population history. Thus, our study contributes to a deeper understanding of genetic diversity and evolutionary processes within the Mustelinae, while also offering valuable guidance for the development of conservation strategies and the preservation of genetic diversity in these species.
Results
Two New Chromosome-level Genome Assemblies
We improved the previously published draft genome assembly of M. nivalis (GCA_019141155.1) (Miranda et al. 2021) to chromosome-level and generated the first chromosome-level assembly for M. strigidorsa. The M. nivalis assembly includes 21 chromosomal scaffolds (Fig. S1a), which correspond to the published karyotype (2n = 42) (Graphodatsky et al. 2020), and chromosome numbering follows this karyotype (Table S2). For M. strigidorsa we observed 2n = 44, similar to M. erminea (Fig. S1b) (Graphodatsky et al. 2020); this difference is explained by a fusion of chr14 and chr16 in M. nivalis (Fig. S1c). A one-to-one synteny between M. strigidorsa and M. erminea chromosome scaffolds was observed (Fig. S1c); thus we decided to use the same chromosome numbering for both species (Table S3).
The genome assemblies of M. strigidorsa and M. nivalis have total lengths of 2.42 Gbp and 2.45 Gbp, respectively (Table S4). Notably, the assembly lengths differ from the genome size estimates based on 23-mers: 3.21 Gbp for M. nivalis and 3.34 Gbp for M. strigidorsa (Table S5). The assembly lengths are comparable to the lengths of other species assemblies included in our study, ranging from 2.41 Gbp for the M. sibirica to 2.68 Gbp for the N. vison. The scaffold N50 s, 115.1 Mbp (M. strigidorsa) and 138.4 Mbp (M. nivalis) are also comparable to the values for other Mustelinae assemblies (130.15–220.3 Mbp). The BUSCO analysis using the Mammalia odb10 dataset (2024 to 2001-08, total BUSCOs = 9226) on the obtained genome assemblies of M. strigidorsa and M. nivalis showed 94.6% and 96% complete and single-copy BUSCOs, with only 3.9% and 2.9% missing BUSCOs, respectively, which is similar to other Mustelinae assemblies (Fig. S2, Table S6).
Genome Size Variation in Mustelinae Species
We observed significant variation of genome sizes across the analyzed species and samples (Fig. 2, Table S5). The estimates ranged from 2.16 Gbp for M. putorius (ERR7260426) to 3.35 Gbp for M. nivalis (T100). The greatest variation was detected within M. putorius and M. richardsonii. The M. putorius samples formed two distinct groups: one ranging from 2.16 Gbp to 2.3 Gbp and another from 2.53 Gbp to 2.63 Gbp. Among M. richardsonii samples, one outlier exhibited a notably small genome size of 2.29 Gbp, while the remaining samples ranged from 2.58 Gbp to 2.79 Gbp. The smallest differences in genome size were observed between N. vison individuals. An analysis of Transposable Elements (TE) in the genome assemblies revealed TE content ranging from 35.85% to 39.6% (Table S7). Most TEs belonged to the SINE and LINE superfamilies. Minimal SINE and LINE content were observed in the M. putorius furo genome with 9.61% and 19.02%, respectively. Kimura distance-based copy divergence profiles showed similar patterns (same number and location of the peaks) for assemblies across the seven species which included three or more individuals (Fig. S3).
Estimation of genome sizes for Mustelinae species based on k-mer analysis. Short horizontal lines within boxplots represent the mean value, and long lines indicate the median value. Species abbreviation: MPUT, Mustela putorius, MPFUR, Mustela putorius furo, MEVE, Mustela eversmanii, MRIC, Mustela richardsonii, NVIS, Neogale vison, MSIB, Mustela sibirica, MNIG, Mustela nigripes, MERM, Mustela erminea, MNIV, Mustela nivalis, MSTR, Mustela strigidorsa. Detailed data are provided in Table S5.
Genome Synteny and Rearrangements
We reconstructed a macro-level synteny map for the available genomes of the Mustelinae as well as other species within the Mustelidae family (Fig. 3). This revealed highly conserved syntenic segments, mostly maintaining centromere positions across species. However, some exceptions indicate evolutionary shifts in centromere positions within the genus Mustela. For example, centromere positions in homologous chromosomes of M. strigidorsa and M. erminea differ from those in other Mustela species (Fig. S4). A centromeric shift was observed for MERM 12 and its homologs (Fig. S4a): MSTR 12 and MERM 12 are acrocentrics, whereas MNIV 13, MLUT 9, MPFUR 10 and MNIG 9 are submetacentrics. A similar pattern transformed subtelocentric MSTR 5 and MERM 5 to acrocentric MNIV 17 and MLUT 13, or even to acrocentric MPFUR 16 and MNIG 13 (chromosome numbers are used throughout). Notably, these shifts occurred without accompanying structural rearrangements (Fig. S4b).
Macro-level synteny map among species within the mustelinae and mustelidae. Chromosomes labeled by primes (′) were reverse complemented. Fusions and fissions are highlighted in blue. Inversions larger than 1 Mbp are highlighted in red. Macro-level synteny (gray lines) is shown between chromosomes (horizontal colored lines) of seven species of Mustelinae, one Lutrinae (ELUT), and one Guloninae (MFOI) species. Centromeric positions are tentative, approximately drawn based on the analysis of comparative chromosome painting maps and G-banded karyotypes. Abbreviation: MFOI, Martes foina, ELUT, Enhydra lutris, NVIS, Neogale vison, MSTR, Mustela strigidorsa, MERM, Mustela erminea, MNIV, Mustela nivalis, MLUT, Mustela lutreola, MPFUR, Mustela putorius furo, MNIG, Mustela nigripes.
Despite these generally conserved patterns, we identified a number of structural rearrangements in Mustela genomes, including new inversions (Fig. 3, red connectors) and all previously described Robertsonian translocations (blue connectors). Some of the inversions are likely lineage-specific. For example, inversions between MNIV 12, 13 and 15, and their homologs MLUT 15, 9 and 10 likely distinguish the ferret-like (MLUT, MPFUR, MNIG) lineage from other Mustela species (MSTR, MERM, MNIV). The sizes of these inversions are approximately 6.89 Mbp, 2.23 Mbp, and 13.19 Mbp. At the same time, NVIS stands out due to numerous translocations and species-specific inversions, even if multiple fissions and fusions of chromosomes are not considered. Expectedly, distinct heterochromatic arms observed in cytogenetic studies on the chromosomes of some Mustela species (Graphodatsky et al. 1976, 1977, 1989, 2002) are absent in current genome assemblies, which is evident from the reconstructed syntenic blocks. For instance, the large heterochromatic arm on MNIV 1 and MERM 9 is missing.
Phylogenomics and Karyotype Evolution of Mustelinae
We first reconstructed a whole-genome phylogeny for the lineage and then integrated these results with aforementioned genome synteny analysis. We generated a ML (Maximum Likelihood) phylogenetic tree using positions that vary between species (including heterozygous ones) from 6,599 single-copy protein-coding genes (Fig. 4a). The inferred topology showed a clear differentiation of the currently recognized species. Just as expected, N. vison appeared as a sister group to the genus Mustela. M. strigidorsa, the only tropical species involved in the analysis, diverged first within Mustela. Next, stoats (M. richardsonii and M. erminea), M. nivalis, M. sibirica, M. lutreola and, finally, the ferret lineage (M. eversmannii, M. nigripes, M. putorius + M. p. furo) split sequentially from the stem. To verify the concatenated ML tree, we reconstructed a coalescent-based phylogeny (Fig. S5), and got a completely congruent topology. All internal nodes at and above the species-level showed high posterior probabilities (pp1 = 1.0). However, quartet support values (q1) varied across the tree (Fig. S6, Table S8). The lowest support was observed for node 9 (Most Recent Common Ancestor (MRCA) of M. sibirica and M. lutreola), with only 47.7% of quartets supporting the main topology (q2 = 30.2%, q3 = 21.9%). A higher, but still reduced, quartet support was detected for node 2 (q1 = 61.7%), which corresponds to the MRCA of the Mustela and Neogale (N. vison). All other interspecific nodes were supported by a high fraction of the quartets (q1 = 72.8% to 88.7%).
Phylogenomic relationships, genetic distances, and ancestral karyotype reconstruction in mustelinae. a) Maximum likelihood phylogram based on nuclear phylogenomic data of analyzed Mustelinae species. Only autosome data were used for the reconstruction, and the tree was rooted on Martes foina (not shown, mfoi.min_150.pseudohap2.1_HiC, DNA Zoo). Labels near branches correspond to ancestral fissions (red) and fusions (green). The translocations were encoded relative to M. strigidorsa chromosomes, as they represent the chromosome-level syntenic blocks. Note a difference in designation between fissions and fusions. For example, fis(MSTR8, MSTR15) means a fission of ancestral chromosome to homologs of MSTR8 and MSTR15, and fus(MSTR14; MSTR16) means a fusion of homologs of MSTR14 and MSTR16 to a new chromosome. b) Intraspecific and interspecific pairwise distances among Mustelinae species. Colored and black horizontal lines within each boxplot represent the mean and median values, respectively. Red dashed lines and arrows indicate the gap between intra- and interspecific distances. Species abbreviation: MEVE – Mustela eversmanii, MNIV – Mustela nivalis, MERM – Mustela erminea, MRIC – Mustela richardsonii, MNIG – Mustela nigripes. c) The chromosomal synteny (scheme) between ancestral karyotypes (AK) of Carnivora, Mustelidae, Lutrinae, Mustelinae and Mustela. Fissions are highlighted in blue. Chromosome numbering for Mustelidae AK and Lutrinae AK is based on that of Martes foina, for Carnivora AK we followed the previously proposed nomenclature (Beklemisheva et al. 2016), for Mustelinae AK we used M. strigidorsa nomenclature with modification for chr 1 and 2, and for Mustela AK it completely corresponds to M. strigidorsa.
To trace the evolutionary history and reconstruct ancestral states of the detected Robertsonian translocations, we encoded them as fissions or fusions relative to the M. strigidorsa genome (Fig. S7), for example, fis(MSTR8, MSTR15) and fus(MSTR8, MSTR13), and mapped them onto the phylogenetic tree (Fig. 4a). We got consistent results with a previous cytogenetic study (Graphodatsky et al. 2002). However, inclusion of the M. strigidorsa assembly, generated in this study, resulted in a better relative placement of the fission event fis(MSTR2; MSTR21). Specifically, while the earlier study suggested that fis(MSTR2; MSTR21) occurred after the divergence of Neogale but before the split of M. erminea, our phylogenetic placement of M. strigidorsa, as a basal lineage within Mustela, indicates that the event likely occurred even earlier, prior to the diversification of extant Mustela species.
Taxonomic Uncertainties Within Mustelinae
Two species within Mustela are both widespread and taxonomically challenging: M. erminea and M. nivalis. Mitochondrial data have indicated that North American M. erminea includes multiple species (Colella et al. 2021), but the reliance on mtDNA is complicated by frequent hybridization among Mustelinae. Although mitochondrial (Fig. S8) and nuclear trees (Fig. 4a) are largely congruent, we detected clear cases of mitonuclear discordance: the mitochondrial genome PQ821906 of M. eversmanii sample ERR11751895 (male) clusters with M. putorius in the mitochondrial tree, and sample ERR7260426, identified as M. putorius (male), carries M. lutreola mtDNA (BK069848). Such discordance limits the significance of mitochondrial data for Mustelinae and emphasizes the need for genome-wide analyses. Given this, we assessed species delimitation in M. erminea using genome-wide data and applied the same approach to M. nivalis, where species-level divergence has not yet been rigorously tested. To test the hypothesis that there is species-level divergence within M. nivalis, we compared genetic distances between and within sister species (Fig. S9): M. nivalis (9.77 × 10^−4^–1.16 × 10^−3^), M. erminea (1.08 × 10^−3^–1.19 × 10^−3^), M. richardsonii (1.47 × 10^−3^–1.66 × 10^−3^), M. eversmanii (5.56 × 10^−4^–9.12 × 10^−4^), M. erminea-M. richardsonii (2.30 × 10^−3^–2.66 × 10^−3^) and M. eversmanii-M. nigripes (2.60 × 10^−3^–2.80 × 10^−3^). We found a clear differentiation between inter- and intraspecific genetic distances (Fig. 4b). Distances within M. nivalis are relatively small and clearly fall within the intraspecific category: its mean (1.05 × 10^−3^) does not significantly differ from the M. erminea mean (1.15 × 10^−3^, two-sided Mann-Whitney test p-value 0.095) and is smaller than the mean for M. richardsonii (1.15 × 10^−3^, one-sided Mann-Whitney test P-value = 0.00068). At the same time, our comparison confirms the previous split of M. richardsonii and M. erminea as the mean distance between them is similar to the distance between M. eversmanii and M. nigripes (2.47 × 10^−3^ vs. 2.72 × 10^−3^).
Genetic Diversity and Demographic Histories
To investigate how past demographic processes have shaped contemporary genomic variation, we integrated analyses of heterozygosity (Fig. 5, Table S9), Runs of Homozygosity (RoH) (Fig. 6, Table S10), and historical effective population size (N_e_) (Fig. 7) inferred with the Pairwise Sequentially Markovian Coalescent (PSMC) model (Li and Durbin 2011). We detected significant variation in mean heterozygosity (SNP/kbp) both among and within species. Taxa sampled across broad geographic ranges showed the largest intraspecific differences: M. putorius (0.26–0.8), M. eversmanii (0.54–0.62), M. sibirica (0.85–1.11), M. richardsonii (1.28–2.94), M. erminea (1.87–2.14), and M. nivalis (2.23–2.9). Species with restricted distributions or a domestication history had uniformly low diversity (M. nigripes 0.02–0.04, M. putorius furo 0.13–0.25, N. vison 0.85–0.86). M. strigidorsa is represented by a single sample with mean heterozygosity of 0.62 SNP/kbp.
Heterozygosity of Mustelinae species. a) Distribution of the heterozygous SNP density on the chromosomes of Mustelinae species. Heterozygous SNPs were counted in 1 Mbp windows with 100 kbp steps and scaled to SNPs/kbp (represented by color scale on the left, ranging from dark blue (extremely low heterozygosity) to brown (very high heterozygosity), with 0 to 0.1 and over 6 heterozygous SNPs per 1 kbp, respectively. Centromeres are not indicated for pseudochromosome-level assemblies where chromosomes are labeled with the prefix “pchr.” Each heatmap represents a single individual selected as a representative of the corresponding species. b) Intra and interspecific heterozygosity among Mustelinae species. The values at the top of the figure reflect the intraspecies mean and median heterozygosity. Short vertical lines within boxplots represent the mean value and long lines indicate the median value. Each dot corresponds to a separate individual. Species abbreviation: MNIG, Mustela nigripes; MPFUR, Mustela putorius furo; MPUT, Mustela putorius; MEVE, Mustela eversmanii; MSTR, Mustela strigidorsa; NVIS, Neogale vison; MSIB, Mustela sibirica; MERM, Mustela erminea; MRIC, Mustela richardsonii; MNIV, Mustela nivalis. Species global conservation status (CS) according to IUCN data: EN, endangered; NE, not evaluated; LC, least concern; N, number of individuals. Detailed data are provided in Table S9. Distribution of heterozygous SNP density on chromosomes for the other samples is presented in the Supplementary File.
The runs of homozygosity (RoH) content in the Mustelinae samples by species. Cumulative distribution plots of RoH. Homozygous tract lengths and cumulative genome fraction in RoH are represented on X and Y axes. Tracts are ordered from shortest to longest. X chromosomes were excluded from all samples. Detailed data are provided in Fig. S10 and Table S10.
Demographic history of Mustelinae species. The X chromosome was excluded from the analysis. Parameters: g, generation time, μ, mutation rate. Abbreviations: MIS, Marine Isotope Stages: 6 (191–130 kya), 7 (243–191 kya), 8 (300–243 kya), 9 (337–300 kya), 10 (374–337 kya), 11 (424–374 kya), 12 (478–424 kya), 13 (533–478 kya), 14 (563–533 kya); MPT, Mid-Pleistocene Transition (1.25–0.7 Mya).
Extensive homozygosity is evident in M. nigripes and M. putorius furo (Fig. 6, Fig. S10, Table S10): M. nigripes RoH cover 2.1–2.2 Gbp (86.83% to 91.66%) out of 2.5 Gbp of the genome assembly with 77.83% to 91.81% in ultra-long tracts (≥10 Mbp), while M. putorius furo had a 1.48–1.88 Gbp fraction (64.9% to 82.05%) of the genome assembly, of which 38.75% to 54.09% were long RoH (≥1 Mbp). M. putorius has the highest variation in total RoH length (371.8 Mbp–1.4 Gbp; 15.84% to 59.28%): RoH in European mainland individuals encompass from 15.84% to 39.96% of the genome assembly (2.5 Gbp) and most are short (<1 Mbp), whereas UK samples have a RoH fraction 49.87% to 52.75% of the genome assembly. Sample ERR7256386 (Spain) stands out from the other samples by its RoH content (1.4 Gbp in total), with a major contribution from ultra-long RoH (60.1%). In M. eversmanii samples 335–631 RoH accounted for 23.47% to 27.84% of the genome assembly, with 57.35% to 77.9% contained in 9–13 ultra-long tracts. Highly heterozygous species (M. richardsonii, M. erminea, M. nivalis) were largely devoid of RoH, aside from isolated long segments. For example, M. nivalis T100 had 270 Mbp (11.6%) and S8606 131 Mbp (5.63%), with T100 including seven ultra-long RoH up to 85 Mbp.
PSMC trajectories were consistent with these diversity patterns. High-diversity species showed large and relatively stable historical N_e_ (Fig. 7). European M. nivalis samples displayed a steady N_e_ increase following divergence from Asian lineages 1 Mya (Mid-Pleistocene Transition (MPT), confidence interval (CI): from 629.6 kya to 1.6 Mya), reaching up to 62.6k. Asian individuals remained lower (up to 20.4k) and did not show recovery. M. erminea exhibited parallel trajectories across samples, with N_e_ rising until 100 kya (CI: 63–157.8 kya, max 22.7–25.1k) before a gradual decline. M. richardsonii and M. erminea diverged 2 Mya (CI: 1.3–3.2 Mya), followed by elevated N_e_ (max 17.5–22.6k) and stabilization near 200 kya (CI: 125.9–315.6 kya). The hybrid M. richardsonii × M. erminea individual (SRR6963887) (Colella et al. 2018, 2021) showed a distinct continuous increase to 32.7k until 150 kya (CI: 94.4–236.7 kya). Species with lower heterozygosity and extensive RoH experienced recurrent or prolonged bottlenecks. M. sibirica showed two contractions (at 350 kya, CI: 220.4–552.4 kya; and at 100 kya), reducing N_e_ from 8.8k to 4.1k. M. eversmanii underwent a sharp decline beginning 1 Mya, reaching minima of 2.9–3.4k. M. strigidorsa exhibited two reductions (850 kya, CI: 535.1 kya–1.3 Mya, to 350 kya; and 150 kya) to a minimum of 2.3k. M. putorius experienced two pronounced bottlenecks (500 kya, CI: 314.8–789.1 kya; and 200 kya), reducing N_e_ to 0.4–1.2k; continental and UK individuals formed two clusters but shared the same decline pattern. In M. nigripes, extremely low genomic diversity restricted inference to 350 kya, with consistently low N_e_ (0.7–1.1k). All N. vison samples showed a prolonged decline beginning 1 Mya, stabilizing around 300 kya (CI: 188.9–473.5 kya).
Discussion
Genome Assembly Length vs. Genome Size
In recent years, chromosome-level genome assemblies have become the gold standard in comparative and population genomics, providing highly detailed genomic structures and facilitating sophisticated genetic studies (Theissinger et al. 2023). Within the Mustelinae, however, such resources remain scarce (Table S1): only six chromosome-level and two scaffold-level assemblies were previously available, with considerable variability in assembly quality due to differences in sequencing technologies. Long-read–based assemblies of M. erminea, M. lutreola and N. vison stand out for high continuity and BUSCO completeness, whereas our new chromosome-level assemblies of M. nivalis and M. strigidorsa as well as the scaffold-level assembly of M. sibirica achieve quality metrics comparable to those of other Mustela genomes (Tables S4 and S6). Despite this progress, genome assemblies for 11 Mustelinae species are still missing, largely due to difficulties in obtaining samples from tropical regions (M. kathiah, M. nudipes, M. lutreolina) and from geographically restricted islands (M. haidarum, M. itatsi). The newly generated M. strigidorsa genome assembly broadens taxonomic representation within tropical Asian Mustelinae and provides a critical reference for future comparative and phylogenomic studies.
According to previous cytogenetic studies, Mustelinae species are often characterized by completely heterochromatic arms (consisting almost exclusively of the additional heterochromatin) on originally acrocentric chromosomes (Graphodatsky et al. 1977, 1989). Depending on lengths, which vary from species to species, they can notably affect the genome sizes (Graphodatsky et al. 1977). These heterochromatin blocks, comprised largely of difficult-to-sequence tandem repeats with high GC content, are often poorly represented in assemblies due to sequencing biases and the discarding of repeat-associated reads during assembly (Biscotti et al. 2015; Sedlazeck et al. 2018). Our data appear to confirm that differences in heterochromatin content underlie the variability in genome size among Mustelinae species, as indicated by discrepancies between estimates derived from genome assemblies and 23-mer distribution of WGS reads (Fig. 2, Table S5). These discrepancies are evident when comparing genome size estimates from different methods, such as k-mer distribution analysis of WGS data (Kliver et al. 2017; Ranallo-Benavidez et al. 2020) and cytogenetic analysis (Hardie et al. 2002). Cytogenetic data suggest the euchromatic portion remains stable across Mustelidae species (2.4 to 2.9 Gbp, which corresponds to 2.5 to 3 pg [Doležel et al. 2003]) (Graphodatsky 1989), with size differences attributed to heterochromatin variation (Graphodatsky et al. 1977). In our study, genome assemblies (2.41–2.68 Gbp, Table S4) and 23-mer WGS-based estimates (Table S5) consistently yielded smaller sizes than cytogenetic measurements. Species with minimal heterochromatin content such as M. sibirica (13.4% ± 4.7% [Graphodatsky et al. 1977]) with an assembly length of 2.4 Gbp showed moderate differences in genome size (cytogenetic: 3.0–3.1 Gbp = 3.1 to 3.2 pg vs. WGS: ∼2.54–2.59 Gbp), while those with substantial heterochromatin like M. nivalis (34.5% ± 3.2% heterochromatin [Graphodatsky et al. 1977]) with an assembly length of 2.45 Gbp exhibited more dramatic disparities (cytogenetic: 4.4 Gbp = 4.5pg vs. WGS: 3.08–3.35 Gbp). In other Mustelinae species, such as M. eversmanii, M. putorius, M. erminea and N. vison, the heterochromatin content is 20.6% ± 2.2%, 22.7% ± 3.4%, 22.8% ± 3.5% and 25.4% ± 4.7%, respectively (Graphodatsky et al. 1977). These inconsistencies likely stem from incomplete assembly of heterochromatic regions in genome sequencing approaches, compounded by the high error margins (up to 15%) in heterochromatin quantification methods (Graphodatsky et al. 1983; Graphodatsky 1989).
Phylogeny and Ancestral Rearrangements
Previous Mustelinae phylogenies were primarily based on concatenated mitochondrial and/or limited nuclear markers (Koepfli et al. 2008a; Sato et al. 2012; Law et al. 2018; Hassanin et al. 2021). We performed such a reconstruction using whole-genome data for the first time, including new genome assembly and resequencing data of two Asian species, M. strigidorsa and M. sibirica (Fig. 4a). M. strigidorsa, which is distributed in the tropical regions of Southeast Asia, occupied a basal position relative to other Mustela species in the phylogenetic trees (Fig. 4a, Figs. S5 and S6). For three more tropical Asian species (M. kathiah, M. nudipes, and M. lutreolina), whole-genome data are still unavailable, but mitochondrial genome reconstructions suggest successive splitting of M. nudipes and M. kathiah from the stem (Hassanin et al. 2021), while concatenated nuclear + mitochondrial DNA datasets group M. nudipes with M. strigidorsa together as sister taxa (Koepfli et al. 2008a; Sato et al. 2012; Law et al. 2018). However, mtDNA trees might be unreliable in case of mustelids, due to active interspecies hybridization within this family, which was reported for multiple species: badgers, martens, ermines, ferrets and others (Rozhnov et al. 2013; Kinoshita et al. 2019; Colella et al. 2021; Szatmári et al. 2021; Etherington et al. 2022; Tomarovsky et al. 2025). Breeding in captivity has also revealed that distantly related species such as M. putorius and M. sibirica are capable of hybridizing (Graphodatsky et al. 1982, 1985). In our study we observed some discordance between nuclear (Fig. 4a) and mitochondrial (Fig. S8) trees. For example, the M. putorius ERR7260426 (BK069848) sample was previously shown to have such a discordance (Etherington et al. 2022); inclusion of all available genome data from the Mustelinae clarified that this sample carries a M. lutreola mitochondrial genome.
Our species tree (Figs. S5 and S6) supports the distinction between N. vison and the genus Mustela, with high main posterior probability support (pp1 = 1.0, Table S8). While quartet support for this split was somewhat lower than for other interspecific nodes (q1 = 61.7%), it remains above the threshold typically considered indicative of primary topological signal. This moderate value may reflect inherent gene tree discordance due to incomplete lineage sorting or ancient gene flow at the early stages of Mustelinae diversification. Nonetheless, both concatenated (Fig. 4a) and coalescent-based (Figs. S5 and S6) analyses consistently recover N. vison as a sister lineage to Mustela, supporting the current taxonomic treatment that recognizes Neogale as a distinct genus. While most interspecific nodes were characterized by high quartet support, a notable exception is node 9, uniting M. sibirica and M. lutreola, which showed markedly reduced support (q1 = 47.7%, Table S8), likely reflecting conflicting gene tree signals in this part of the phylogeny. At the same time, the position of M. strigidorsa as the earliest branching lineage within Mustela is strongly supported, further reinforcing its placement within this genus.
M. erminea sensu lato (all stoats) and M. nivalis are the Mustelinae species with the largest ranges, encompassing nearly the whole Holarctic (Fig. 1). The combination of such a wide distribution and limited dispersal due to small body sizes suggested to researchers that these two taxa might contain multiple species. Recently, M. erminea sensu lato was split into three species: *M. erminea (*sensu stricto, Eurasia and Alaska), M. richardsonii (continental America), and M. haidarum (Haida Gwaii and Alexander Archipelago of coastal North America) (Colella et al. 2021). However, this suggestion was based on mitochondrial sequences and morphological traits only. By combining samples from a previous dataset (Colella et al. 2018) and sequencing two more M. erminea individuals from Yakutia, Russia, we confirmed the hypothesis of multiple species (Supplementary Results, Discussion, and Methods). Due to the lack of samples, the status of European M. erminea remains unclear. For M. nivalis we found that despite clear division of Western European and Asian samples, pairwise distances between them are comparable to distances within M. erminea (Fig. 4b, Fig. S9), i.e. no signs of speciation were observed. However, it is important to note that within-species divergence was assessed for Eurasian M. nivalis only, due to the lack of samples from North America.
The order Carnivora was suggested to have an ancestral karyotype with 2n = 38 (Nash et al. 2008; Perelman et al. 2012; Beklemisheva et al. 2016), which is observed across a wide range of terrestrial and semiaquatic species (Graphodatsky et al. 2020). Within Carnivora, the Mustelidae also exhibits a predominantly syntenic conservatism with 2n = 38 in most species (Graphodatsky et al. 2020). However, several lineages within Mustelidae show notable variation in chromosome number, for example, Melinae (e.g. Meles, 2n = 44) and Guloninae (e.g. Martes, 2n = 38–40; Gulo, 2n = 42), reflecting independent chromosomal rearrangements (Graphodatsky et al. 2020). One such example is the Mustelinae, which displays considerable karyotypic variability (Figs. 3 and 4a). Within this group, the diploid number ranges from 2n = 30 in N. vison to 2n = 44 in species such as M. strigidorsa, M. kathiah (Abramov et al. 2013), M. erminea, and M. altaica (Graphodatsky et al. 2020). This variability likely reflects a history of recurrent chromosomal fissions and fusions, which have contributed significantly to the evolutionary diversification of the subfamily. The results of our study allow us to clarify the timing of the three latest chromosomal fissions in Mustelinae (Fig. 4c). All three rearrangements occurred after the divergence of Mustelinae from other Mustelidae lineages and at early stages of the subfamily's radiation (Fig. S7). These events were previously identified (Graphodatsky et al. 2002), but two of them are now confirmed to have taken place before the diversification of extant Mustelinae, while the timing of the third fission has been revised based on our data. Earlier hypotheses suggested that the fission fis(MSTR2, MSTR21) occurred after the divergence of the Neogale lineage but before the separation of M. erminea (Graphodatsky et al. 2002). The inclusion of M. strigidorsa in our phylogenetic analysis provides important new evidence. This species occupies a basal position within the Mustela clade (Fig. 4a), which shifts the previously proposed timing of the fission event. Additional support for the ancestral Mustelinae rearrangements comes from data on other tropical Mustela species. For example, M. kathiah has been shown in previous studies (Sato et al. 2012; Law et al. 2018), as well as in our mitochondrial genome analysis (Fig. S8), to have diverged after M. strigidorsa. Furthermore, M. nudipes is phylogenetically close to M. strigidorsa (Koepfli et al. 2008a; Sato et al. 2012; Law et al. 2018), and M. lutreolina is another tropical species potentially related to this group. However, the phylogenetic position of M. lutreolina remains uncertain, as no cytogenetic or genomic data are currently available for this species. These observations support the inference that the ancestral Mustela karyotype most likely had a diploid number of 2n = 42 (Fig. 4c). The ancestral karyotype of the genus Neogale remains uncertain. In N. vison, several chromosomal fusions have been identified, resulting in a reduced chromosome number of 2n = 30 (Fig. 3). However, it is not yet clear whether these chromosomal rearrangements are specific to this species or represent a broader pattern shared across the genus. Further studies, including data from other Neogale species are necessary to address this question.
Genetic Diversity and Demographic Histories
Maintaining genome-wide genetic variation is widely considered critical for population viability (Lande and Shannon 1996; Abascal et al. 2016; Bozzuto et al. 2019), while some argue for focusing on functionally important variation (Robinson et al. 2016, 2019; Teixeira and Huber 2021). Despite ongoing debates, there is broad consensus that reduced genome-wide diversity can affect long-term viability, despite context-dependent effects. Mustelinae species, like others, are subject to varying climatic and ecological pressures, leading to significant fluctuations in their genetic diversity (Colella et al. 2018; de Ferran et al. 2022; Derežanin et al. 2022). In our study, we evaluated genetic diversity by assessing and comparing genome-wide heterozygosity and homozygosity within and between species (Figs. 5 and 6), as well as investigating their distribution within the genomes. In addition, our demographic analysis revealed significant variation in effective population size among Mustela species (Fig. 7), reflecting biogeographic history and responses to Pleistocene climate change. Although our data do not cover population-level resolution, they provide an initial genome-wide overview and demonstrate shared impacts of large-scale environmental factors.
We found considerable intraspecific and interspecific differences in mean heterozygosity between analyzed Mustela species (Table S9): from 0.02 SNPs/kbp to 2.9 SNPs/kbp. The most heterozygous species included the widely distributed M. nivalis (2.64 SNP/kbp), M. erminea (2.03 SNP/kbp) and M. richardsonii (2.22 SNP/kbp). The observed high genetic diversity in M. nivalis samples aligns with previous studies based on different molecular markers, including mitochondrial sequences (Lebarbenchon et al. 2010; Sato et al. 2020; Tissaoui et al. 2024). However, despite the high mean heterozygosity of this species, we observed clear signs of recent inbreeding, indicated by the presence of ultra-long RoH (e.g. in sample T100, Table S10) covering significant portions of their chromosomes. This is consistent with monitoring studies that show declines in local populations of M. nivalis in regions such as the USA (Jachowski et al. 2021), UK (Sainsbury et al. 2019), Spain (Llorca et al. 2024) and Tunisia (Hayder et al. 2023).
A combination of high heterozygosity and broad ecological tolerance in M. nivalis and M. erminea (Sommer and Crees 2022) likely promoted their demographic stability during climatic oscillations. Both show either continuous growth or consistently elevated N_e_ during MIS (Marine Isotope Stage) 14–6 (Fig. 7), a period of strong climatic instability. These patterns are consistent with their persistence across glacial–interglacial cycles and broad geographic ranges (Sommer and Benecke 2004; Sommer and Crees 2022), a notion further supported by fossil evidence (Sommer and Benecke 2004; Marciszak and Socha 2014; Krajcarz et al. 2015; Kosintsev et al. 2016; Crégut-Bonnoure et al. 2018; Giustini et al. 2024). A more detailed look reveals a structured demographic history for M. nivalis, with western (European) and eastern (Asian) lineages diverging around 1 Mya during the MPT, followed by sustained N_e_ growth in the west and relative stability in the east. We hypothesize that the elevated N_e_ in Europe may indicate historical gene flow, a pattern similar to that observed in the hybrid M. richardsonii × M. erminea individual (SRR6963887), where introgressive hybridization is well documented (Colella et al. 2018). Such complex introgression signatures, also observed in other Mustelidae hybrids like M. zibellina × M. martes (Tomarovsky et al. 2025), can be indirectly detected by PSMC, which, while not explicitly inferring admixture, offers indirect evidence of complex histories. The demographic stability in eastern M. nivalis also has a broad range overlapping. distributed taxon but has been relatively understudied. Its intraspecific structure remains unknown and the species is still treated as a single taxonomic unit. Consequently, there is reason to suggest that M. nivalis may comprise distinct, geographically structured lineages that could warrant species-level recognition consistent with a possible Asian origin and later westward expansion. M. erminea shows a broadly similar trajectory, with N_e_ increase during MIS 14–6 followed by decline near the end of MIS 6. However, due to sampling from eastern populations only, spatial inferences are limited. Nevertheless, the concordant demographic patterns observed in M. nivalis and M. erminea support the hypothesis that their persistence was shaped by shared ecological mechanisms, particularly their high adaptability to cold climates during periods of Quaternary climatic instability.
In stark contrast to these widespread species, we observed significantly lower heterozygosity and extensive RoH in the less widespread Mustela species (Tables S9 and S10), which is consistent with evidence of recurrent or prolonged bottlenecks (Fig. 7). A notable example is the tropical M. strigidorsa, the species previously unassessed on the genome level. The single sample from Vietnam has a mean heterozygosity of only 0.62 SNP/kbp (Fig. 8a), with RoH covering 23.62% of its genome. While it exhibits relatively moderate homozygosity, its genome shows both long (10.21%) and ultra-long (1.81%) RoH. Although there are concerns regarding habitat degradation and fragmentation for this species, their effect on its population size remains unknown due to limited data (Abramov et al. 2008). Its genetic diversity is significantly lower than that of another South Asian species with endangered status, Ailurus fulgens (Fig. 8a). Furthermore, similarly with M. eversmanii, it occupies an intermediate position between the highly heterozygous Mustelinae and species with well-documented threatened status, including Enhydra lutris, Lutra lutra, Acinonyx jubatus, and Neofelis nebulosa (Fig. 8a). At the extreme end of the homozygosity spectrum is M. nigripes, a species once declared extinct (Fricke 2015), where RoH cover at least 87% of the genome, and heterozygosity ranges from 0.02 to 0.04 SNP/kbp (Derežanin et al. 2025)—significantly lower than that of the domesticated M. putorius furo.
Distribution of the heterozygous SNP density in Mustelinae species and five well-known in conservation genetics species: Enhydra lutris, Lutra, Acinonyx jubatus, Neofelis nebulosa, and Ailurus fulgens (Totikov et al. 2021), and the status of main fur game species in Russia. a) Heterozygous SNPs were counted in 1 Mbp windows and 100 kbp steps and scaled SNPs/kbp for all species. Only samples with highest mean values are shown (one sample per species, Table S9). Median and mean heterozygosity values are indicated within boxplots by black line and white dots, respectively. Color gradient indicates the level of heterozygosity, ranging from lower (blue) to higher (red) values. b) The main fur game animal resources in the Russian Federation for the years 2003 to 2023. Data are based on public reports of the Ministry of Natural Resources and Environment of the Russian Federation and the Federal Research Center for Hunting Development (Lomanov et al. 2007; Minprirody of Russia 2024). Dotted lines indicate discrepancies in the datasets (Table S11). Abbreviations: GS, Global conservation status according to IUCN Red List data; LC, Least Concern; NT, Near Threatened; VU, Vulnerable; EN, Endangered; NE, not evaluated; Species: MNIG, Mustela nigripes; ELUT, Enhydra lutris; LLUT, Lutra; MPFUR, Mustela putorius furo; AJUB, Acinonyx jubatus; NNEB, Neofelis nebulosa; MEVE, Mustela eversmanii; MSTR, Mustela strigidorsa; MPUT, Mustela putorius; NVIS, Neogale vison; AFUL, Ailurus fulgens; MSIB, Mustela sibirica; MERM, Mustela erminea; MRIC, Mustela richardsonii; MNIV, Mustela nivalis; MMAR, Martes; MFOI, Martes foina; GGUL, Gulo; MZIB, Martes zibellina.
The pattern of low diversity extends to M. eversmanii (Fig. 5), represented in our study by three samples from its Asian range, which exhibit relatively low mean heterozygosities (0.54, 0.58, and 0.62 SNPs/kbp). This species is considered to be rapidly disappearing in several European countries (Šálek et al. 2013), facing severe threats including habitat loss and fragmentation, declining prey base, competition, and rodenticide poisoning (Šálek et al. 2013; Szapu et al. 2024). Conversely, a recent study based on low-resolution data challenges the alarming rate of decline in Europe, suggesting either insufficient research or a rapid population recovery following a sharp decline (Szatmári et al. 2021). Assessments in its Asian range are scarce, but reports indicate a significant decline in Russia (Lomanov et al. 2007; Kiseleva 2024; Minprirody of Russia 2024). There, the combined population estimate of M. eversmanii and M. putorius (due to nearly indistinguishable tracks) dropped from 90.6 to 48.3k individuals between 2003 and 2023 (Fig. 8b, Table S11), with two species believed to contribute approximately equally to these counts (Lomanov et al. 2007; Minprirody of Russia 2024). Although the causes of M. eversmanii decline in Russia remain unclear, current evidence suggests contributions from competition with the invasive N. vison (Macdonald and Harrington 2003; Croose et al. 2018; Kiseleva 2024), long-term declines of its key prey Spermophilus erythrogenys due to extensive control programs (Shilova 2011; Vazhov et al. 2016; Wright et al. 2022), and broader threats across its range, while habitat loss and hunting pressure appear minor (detailed data are provided in Supplementary Results, Discussion, and Methods) (Kiseleva 2024). A similar situation is observed in M. putorius, which also shows low genetic diversity (0.43 SNPs/kbp) and high RoH (15.84% to 59.28%). Populations are declining in Europe due to factors overlapping with M. eversmanii, such as competition with invasive predators (Croose et al. 2018). Notably, while hybridization with M. putorius furo has contributed to population recovery in the UK (Shaw et al. 2025), increasing competition with N. vison remains a threat (Croose et al. 2018).
Unsurprisingly, given their low genetic diversity and current threats, the demographic histories of these less widespread species show evidence of recurrent or prolonged bottlenecks. M. putorius, widespread in Europe, experienced two sharp N_e_ declines during MIS 12 and MIS 6 (Fig. 7). Despite this, it is considered an early post-LGM (Last Glacial Maximum) recolonizer of Central Europe, supported by fossil data (Sommer and Benecke 2004; Sommer and Crees 2022). Other species, sampled from Asia, show bottlenecks around the onset of the 100-kyr glacial cycles. Although Asia is thought to have been less impacted by Quaternary climate shifts (Fu and Wen 2023), our results suggest substantial demographic changes even in steppe and tropical-adapted species. Fossil data for these taxa remain limited (Krajcarz et al. 2015; Kosintsev et al. 2016; Giustini et al. 2024; Fourvel et al. 2025): M. eversmanii is known from seven European Pleistocene sites (including M. stromeri, its common ancestor with M. putorius and M. nigripes); M. sibirica from two Japanese finds; and M. strigidorsa lacks confirmed Pleistocene records (Peters and McClennen 2016). Southern Polish (Krajcarz et al. 2015) and Ural (Kosintsev et al. 2016) fossils suggest a broader range for M. eversmanii during the Pleistocene, later restricted to Eastern European steppes.
Overall, Mustela demographic histories highlight how long-term population dynamics reflect the interplay of environmental change and climatic history. Future studies incorporating broader genomic sampling, paleodistribution models, and molecular dating are crucial to provide a more comprehensive overview.
Conclusions and Perspectives
This study presents the first comprehensive whole-genome analysis of the subfamily Mustelinae, in which we examined chromosome-scale structural rearrangements, reconstructed phylogenetic relationships, assessed genome-wide genetic diversity, and inferred demographic history for 10 species. Our results show that genetic variability within Mustelinae is shaped by multiple demographic and ecological processes rather than by uniform evolutionary pressures. The pronounced interspecific and intraspecific heterogeneity in heterozygosity highlights the complex evolutionary dynamics operating within this group. Particular attention is drawn to M. eversmanii and M. putorius. Both species show low levels of genetic diversity, RoH tracks, and genomic signals of historical N_e_ reductions. For M. eversmanii, these patterns are evident across both the European and Asian parts of its range, suggesting that the species may be more sensitive to environmental and anthropogenic pressures than previously assumed. Similar genomic signals in M. putorius raise concerns that its current classification as Least Concern may not completely capture its real conservation status. Although our results do not, by themselves, justify IUCN Red List reclassification, they indicate that a more detailed evaluation of genetic and demographic trends would be valuable for refining conservation priorities. Overall, the findings emphasize the need for regular monitoring of genetic diversity in Mustela species, especially in taxa with ongoing population declines and potentially reduced efficacy of purifying selection.
Our phylogenomic reconstruction, the first based on whole-genome data for Mustelinae, refines the evolutionary relationships within the subfamily and supports several key taxonomic conclusions. The results confirm the validity of the genus Neogale, support the placement of M. strigidorsa within the genus Mustela, and validate the species status of M. richardsonii. Nuclear and mitochondrial datasets produced largely congruent topologies, but several discrepancies were detected, likely caused by interspecific hybridization. The first genome of M. strigidorsa, the representative of the tropical and basal Mustela species, allowed us to reconstruct the earlier steps of the Mustelinae karyotype evolution and date the major chromosomal fusions and fissions. We confirmed the previously suggested hypothesis of an ancestral karyotype of 2n = 44 for Mustela and 2n = 42 for Mustelinae. However, the chromosomal evolution within the tropical lineage remains mysterious and represents a promising direction for future research.
Despite the substantial amount of new genomic information generated in this study, many questions remain open, largely because biological samples are difficult to obtain for species with limited or hard-to-access ranges. Future research will benefit from expanding geographic sampling, including unsequenced species, and integrating additional population-level data. Such efforts will allow a more complete understanding of genetic structure, refine models of chromosomal evolution, and support evidence-based strategies for the long-term conservation of mustelid biodiversity.
Materials and Methods
Samples, DNA Extraction and Sequencing
To generate data for the chromosome-level genome assemblies, we used a primary fibroblast cell lines from male M. strigidorsa (MSTR1m, AVA 19-021, origin: Quang Nam, Vietnam) and from male M. nivalis (MNIV1m, origin: Novosibirsk, Russia). The cell lines were provided by the large-scale research facilities “Cryobank of cell cultures,” Institute of Molecular and Cellular Biology SB RAS. We used frozen muscle tissue from a male M. sibirica (MSIB1m, origin: Sakha Republic, Russia) to generate a scaffold-level genome assembly. For whole-genome resequencing of M. nivalis, M. erminea, M. sibirica, and M. putorius, we used both muscle tissue and primary fibroblast cell lines. The resulting dataset, combined with publicly available data, consisted of 50 whole-genome samples representing 10 species from the Mustelinae. A detailed description of the resequencing data is provided in Table S12 and includes both previously published and new data generated within our study.
DNA extraction was performed using the standard phenol–chloroform protocol (Sambrook and Russell 2006). The extracted DNA was fragmented using a Covaris instrument to reach the desired insert size and libraries were constructed with the TruSeq DNA PCR-Free kit (Illumina, Inc., San Diego, CA, USA). For the chromosome-level genome assemblies, we generated Hi-C libraries following the original protocol (Rao et al. 2014). All prepared libraries were sequenced with paired-end 150 bp reads on the Illumina NovaSeq 6000 platform. An Oxford Nanopore Technologies library was also prepared for the M. strigidorsa sample using the SQK-ULK114 kit and sequenced on a 10.4.1 PromethION flow cell with triple loading.
Assembly of the M. Strigidorsa, M. Nivalis, and M. Sibirica Genomes
We assembled the M. strigidorsa genome using Oxford Nanopore long reads (SRR30615031), generated as part of this study, and Hi-C (SRR34091397) and Illumina libraries (SRR34068022), generated by the DNA Zoo Consortium (Dudchenko et al. 2017). We removed adapters from the Oxford Nanopore data using Porechop v0.5.0 with ab initio adapter detection turned off, as in the test runs with the “–ab_initio” option, we detected trimming of mustelid-specific sequences present in the genome assembled using different technologies (Wick et al. 2017). Next, we filtered the data by quality (only reads with mean quality >7 were retained) and trimmed 30 bp from each end using the Chopper v0.8.0 tool (De Coster and Rademakers 2023). First, an initial draft assembly was generated from a single paired PCR-free Illumina library using w2rap-contigger (Clavijo et al. 2017), see (Dudchenko et al. 2018) for details. On the second step, the generated assembly was scaffolded using the SAMBA (script samba.sh with “-m 2500” option) tool (Zimin and Salzberg 2022) from MaSuRCA package v 4.1.0 (Zimin et al. 2013) and the filtered Nanopore data. Errors introduced by the long reads were corrected using the POLCA (from the same version of MaSuRCA) polisher (Zimin and Salzberg 2020) and the Illumina data. Next, we aligned the Hi-C data to the corrected intermediate assembly using Juicer v1.6 (Durand et al. 2016), followed by scaffolding to chromosome-level using 3D-DNA pipeline v210623 (Dudchenko et al. 2017) with subsequent manual curation in Juicebox v2.16.00 (Dudchenko et al. 2018). Finally, we closed gaps using the SAMBA script close_scaffold_gaps.sh (with option “-m 2500”) and the Oxford Nanopore reads and performed a second and third round of polishing.
We upgraded the publicly available draft genome assembly of M. nivalis (GCA_019141155.1), originally generated from linked-read Illumina sequencing data (Miranda et al. 2021), to chromosome-level using the Hi–C sequencing data (SRR34082581), generated by DNA Zoo Consortium. Hi–C data were aligned to the draft genome assembly using Juicer v1.6. Before alignment, the restriction sites for Csp6I + MseI were generated using generate_site_positions.py script in Juicer. Haplotype duplications in the alignments were identified using purge_dups v1.2.6 (Guan et al. 2020), based on sequence similarity and coverage. Regions where more than 90% of the sequence was involved in haplotype duplications were removed. Each deduplicated alignment was then processed using the 3D-DNA v210623 with the default parameters. A manual curation was performed using Juicebox v2.16.00.
To generate a draft genome assembly for M. sibirica, we used w2rap-contigger with the default parameters to assemble paired-end Illumina reads from sample E19. Subsequently, this assembly was scaffolded using the M erminea genome assembly (GCF_009829155.1) as a reference and RagTag v2.1.0 (Alonge et al. 2022) with default parameters.
Multiple Whole-genome Alignment, Synteny Blocks and Localization of Centromeres
Only the chromosomal-level genome assemblies (Table S3 in Supplementary Results, Discussion, and Methods) were used for synteny block analysis. First, we obtained a Maximum Likelihood (ML) phylogenetic tree using BuscoClade v1.7 pipeline (https://github.com/tomarovsky/BuscoClade) with default settings. Second, tandemly organized and dispersed repeats in the genome assemblies were identified and softmasked using Tandem Repeats Finder v4.09.1 (Benson 1999) with parameters “2 7 7 80 10 50 2000 -l 10”, WindowMasker v1.0.0 (Morgulis et al. 2006) with default parameters, RepeatMasker v4.1.6 (Tarailo-Graovac and Chen 2009) with the parameter “-species carnivora”, and BEDTools v2.31.0 (Quinlan and Hall 2010). Next, multiple WGA of all masked assemblies was performed using the obtained phylogenetic tree and Progressive Cactus v2.8.0 (Armstrong et al. 2020) with default parameters. Finally, synteny blocks were extracted from the resulting multiple alignment using halSynteny v2.2 (Krasheninnikova et al. 2020) with parameters “–minBlockSize 50000 –maxAnchorDistance 50000.” The results were visualized using the script draw_macrosynteny.py from the MACE v1.1.32 package (https://github.com/mahajrod/MACE), with parameter “–min_len_threshold 1000000” to highlight only inversions and translocations of 1 Mbp or longer. Centromeres in the analyzed genome assemblies were localized on chromosomal scaffolds by comparison of the corresponding FISH maps and the synteny blocks. The Canis familiaris (UU_Cfam_GSD_1.0, [Hoeppner et al. 2014]) and Homo sapiens (GRCh38.p14, [Schneider et al. 2017]) genome assemblies were used as references. The detailed algorithm for identification of centromere coordinates is described in Kliver et al. (2025).
Read Filtering and Genome Size Estimation
Raw reads were trimmed from adapters and filtered by quality in various ways depending on the sequencing technology (Supplementary Results, Discussion, and Methods). Quality control of the raw and filtered reads was performed using FastQC v0.12.0 (Andrews 2010) and KrATER v2.5 (Kliver et al. 2017). Next, we counted 23-mers from the filtered reads using Jellyfish v2.3.0 (Marçais and Kingsford 2011) with the parameters “-m 23 -s 30G” for jellyfish count and “-l 1 -h 100000000” for jellyfish histo, and then visualized its distribution using KrATER with parameters “-m 23 -u 1.” Finally, the genome size estimation was performed using GenomeScope v2.0 (Ranallo-Benavidez et al. 2020) in diploid mode.
Read Mapping, Variant Calling and Runs of Homozygosity
The filtered reads were first downsampled (Supplementary Results, Discussion, and Methods) to reach ∼12x coverage in all samples. Next, reads from each sample were mapped to the genome assemblies corresponding to its species using BWA v0.7.17-r1188 (Li and Durbin 2009) with the default parameters. The only exception is M. richardsonii, for which no reference genome assembly is currently available; reads from M. richardsonii samples were therefore mapped to the M. erminea reference genome. The Samtools package v1.18 (Li et al. 2009) was then used to sort and mark duplicates in the obtained alignments. The genome coverage in the resulting alignment files was calculated using Mosdepth v0.3.3 (Pedersen and Quinlan 2018) with default parameters.
SNPs and short indels were identified using BCFtools v1.18 (Li 2011) with the following parameters: “-d 250 -q 30 -Q 30 –adjust-MQ 50 -a AD,INFO/AD,ADF,INFO/ADF,ADR,INFO/ADR,DP,SP,SCR,INFO/SCR -O u” for the bcftools mpileup and “–ploidy-file –samples-file –group-samples - -m -O u -v -f GQ,GP” for the bcftools call. The PAR coordinates (Supplementary Results, Discussion, and Methods) were specified for variant calling via the “–ploidy-file” parameter. Low-quality genetic variants were filtered out using the bcftools filter with the parameters “-S. -O z –exclude “QUAL < 20.0 || (FORMAT/SP > 60.0 | FORMAT/DP < 5.0 | FORMAT/GQ < 20.0).” Based on the coverage assessments obtained from the Mosdepth analyses, individual masks for each sample were created using the generate_mask_from_coverage_bed.py script from the MAVR v0.97 with the parameters “-x 2.5 -n 0.33”. These masks were used to remove genetic variants if coverage exceeded 250% or was less than 33% of the median genome coverage. The genetic variants were masked using the BEDTools v2.31.0 with the default parameters. The filtered and masked genetic variants for each sample were divided into heterozygous and homozygous single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) using the bcftools filter with the parameters “-i “TYPE=“snp”'”, “-i “TYPE=“indel”'”, “-i “FMT/GT=“het”'” and “-i “FMT/GT=“hom”'”. Heterozygous SNPs were counted in 1 Mbp windows and 100 kbp steps.
RoH were identified based on the previously calculated heterozygous SNP density in 100 kbp windows with 10 kbp steps using the get_ROH_regions.py script from the Biocrutch v1.0. The algorithm is detailed in (Tomarovsky et al. 2025). RoH coordinates for each sample were determined across all autosomes. The visualization of RoH on the chromosomes was performed using the draw_features.py script from MACE v1.1.32.
Phylogenetic Analyses
We reconstructed genome assemblies for all the resequenced samples based on the reference genome assemblies of the studied species and SNPs. Indels were not taken into account for this analysis. We used the FastaAlternateReferenceMaker tool from the GATK package v4.4.0.0 (Van der Auwera et al. 2013) with the parameter “–use-iupac-sample” to account for heterozygous SNPs. The obtained assemblies and the reference genome assemblies were used to identify single-copy orthologs (6,599) using BUSCO v5.6.1 with the mammalia_odb10.2024-01-08 OrthoDB v.10.1 database. To construct phylogenetic trees, we prepared two datasets: (i) a complete dataset of all resequenced samples and genome assemblies; (ii) a dataset excluding all identified hybrids (see “Admixture analysis of stoat samples” in Supplementary Results, Discussion, and Methods), genome assemblies, and sex chromosomes. Multiple sequence alignment was performed using MAFFT v7.490 (Kuraku et al. 2013), followed by alignment correction by the filtering of hypervariable and poorly aligned regions using Gblocks v0.91b (Castresana 2000). The multiple sequence alignment length after filtering was 11,067,417 bp. Phylogenetic trees were constructed using the ML method implemented in RAxML-NG v1.2.2 (Kozlov et al. 2024) using the GTGTR4 model and 1000 bootstrap replicates. An alternative phylogenetic reconstruction was performed using ASTRAL-III v5.7.1 (Zhang et al. 2018), based on a set of separately reconstructed gene trees using RAxML-NG. To reduce noise, we applied a filtering step by collapsing nodes with bootstrap support below 70% prior to species tree inference. The resulting phylogenetic trees were visualized using the ETE Toolkit v3.1.2 (Huerta-Cepas et al. 2016). All trees were rooted using the Martes foina genome assembly (mfoi.min_150.pseudohap2.1_HiC, DNA Zoo). For reproducibility of the analysis, we employed the BuscoClade v1.7. The genetic distance matrix was obtained using the Neighbor Joining (NJ) method implemented in RapidNJ v2.3.3 (Simonsen et al. 2008).
Ancestral Reconstruction of Chromosomal Rearrangements
For translocations, we created a two-state, presence (Y) or absence (N), trait matrix with species IDs as columns, and rearrangement IDs as rows. Ancestral reconstruction for the internal nodes of the inferred maximum likelihood tree from the trait matrices was performed using the continuous time Markov model implemented in the ape v5.8 package (function rerootingMethod with model=“SYM”) (Paradis and Schliep 2019). Depending on the predicted probabilities (PN) for “N”, we classified states at internal nodes as “N” (PN ≥ 0.7), “Y” (PN ≤ 0.3), and “U” (unclear, 0.3 < PN < 0.7). Our coordinate system was based on the genome assembly of M. strigidorsa (MSTR), which was chosen because this species occupies a basal position in the phylogenetic tree relative to all other analyzed Mustela species. Moreover, it provides the most convenient coordinate framework, as its chromosomes are involved only in fusion events but not in fissions relative to other species. Therefore, for each trait, we checked if the ancestral reconstruction showed “Y” for the root node. In such cases, we invert the state of the trait (from “Y” to “N” and vice versa) for both internal nodes and leaves.
Demographic History
Demographic history was reconstructed using the PSMC v0.6.5 (Li and Durbin 2011) software package with the parameters “-N25 -t15 -r5 -b -p “4 + 25 × 2 + 4 + 6'” applied to data sets both including and excluding the sex chromosomes. Genetic variants were identified using an earlier version of SAMtools v0.1.19, with the alignment quality parameter “-C 50” for samtools mpileup and the variant calling parameter “-c” for bcftools view. Diploid consensus sequences were generated using vcfutils.pl vcf2fq from SAMtools, specifying the minimum (“-d”) and maximum (“-D”) coverage values, calculated for each sample as follows: the median genome coverage divided by 3 for the “-d” parameter and the median genome coverage multiplied by 2.5 for the “-D” parameter. Coverage values outside these ranges were filtered out. Fasta-like sequences were created using fq2psmcfa with a minimum nucleotide quality parameter of “-q20”. Initial bootstrapping, using splitfa, divided the sequences into shorter segments, with 100 rounds of bootstrapping specified by the “-b” parameter for each sample. The files were prepared for visualization using psmc_plot.pl with “-R” parameter. Starting from values reported for the Mustelinae included in the IUCN Red List of Threatened Species (Table S1) (IUCN 2025), generation times (“-g”) were slightly adjusted: 3 years were set for the relatively smaller species (M. nivalis, M. erminea, and M. richardsonii) and 4 years for the larger ones (M. putorius, M. sibirica, M. strigidorsa, M. nigripes, M. eversmanii, and N. vison). A mutation rate (“-u”) of 4.64 × 10⁻⁹ substitutions per generation was applied, with lower (2.94 × 10⁻⁹) and upper (7.37 × 10⁻⁹) confidence interval derived from previous estimates of the germline mutation rate in N. vison (Bergeron et al. 2023).
Supplementary Material
evag014_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abascal F, et al Extreme genomic erosion after recurrent demographic bottlenecks in the highly endangered Iberian lynx. Genome Biol. 2016:17:251. 10.1186/s 13059-016-1090-1.27964752 PMC 5155386 · doi ↗ · pubmed ↗
- 2Abramov A . The taxonomic status of the Japanese weasel, Mustela itatsi (Carnivora, Mustelidae). Zool Zhurnal. 2000:79:80–88.
- 3Abramov AV, Duckworth JW, Wang YX, Roberton SI. The stripe-backed weasel Mustela strigidorsa: taxonomy, ecology, distribution and status. Mammal Rev. 2008:38:247–266. 10.1111/j.1365-2907.2008.00115.x. · doi ↗
- 4Abramov AV, Meschersky IG, Aniskin VM, Rozhnov VV. The mountain weasel Mustela kathiah (Carnivora: Mustelidae): molecular and karyological data. Biol Bull. 2013:40:52–60. 10.1134/S 1062359013010020.23662463 · doi ↗ · pubmed ↗
- 5Alonge M, et al Automated assembly scaffolding using Rag Tag elevates a new tomato system for high-throughput genome editing. Genome Biol. 2022:23:258. 10.1186/s 13059-022-02823-7.36522651 PMC 9753292 · doi ↗ · pubmed ↗
- 6Andrews S . Fast QC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute; 2010.
- 7Armstrong J, et al Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature. 2020:587:246–251. 10.1038/s 41586-020-2871-y.33177663 PMC 7673649 · doi ↗ · pubmed ↗
- 8Beklemisheva VR, et al The ancestral carnivore karyotype as substantiated by comparative chromosome painting of three pinnipeds, the Walrus, the Steller Sea Lion and the Baikal Seal (Pinnipedia, Carnivora). P Lo S One. 2016:11:e 0147647. 10.1371/journal.pone.0147647.26821159 PMC 4731086 · doi ↗ · pubmed ↗