Exploring the gut microbiome and metabolomic interactions of antimetabolite drugs to optimize therapy
Jingyang Chen, Yanan Wang, Lei Xu, Xiaona Li, Libo Zhao

TL;DR
This paper reviews how gut microbes interact with cancer drugs called antimetabolites, aiming to improve treatment outcomes by understanding these interactions.
Contribution
The paper provides a comprehensive framework for assessing chemotherapy-microbiota interactions and identifies potential microbial biomarkers.
Findings
Common antimetabolites affect gut microbial communities in complex ways.
Microbial biomarkers may predict treatment responses to antimetabolite drugs.
Advances in metagenomics and real-time monitoring are needed to optimize therapy.
Abstract
Antimetabolite drugs are cornerstones in treating various cancers and autoimmune diseases; however, their clinical utility is often hampered by systemic toxicity caused by drug-induced gut microbiota dysbiosis. Predicting patient responses remains a significant challenge. Several studies have highlighted the influence of gut microbiota on antimetabolite treatment outcomes, revealing complex bidirectional interactions between the drugs and microbial communities. This review synthesizes the effects of common antimetabolites (including 5-fluorouracil, methotrexate, gemcitabine, capecitabine, 6-mercaptopurine, and thioguanine) on gut microbial communities and outlines a framework (pharmacokinetics, endogenous metabolite production, immune modulation, and apoptotic pathway modulation) for assessing chemotherapy-microbiota interactions. Additionally, potential microbial biomarkers for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1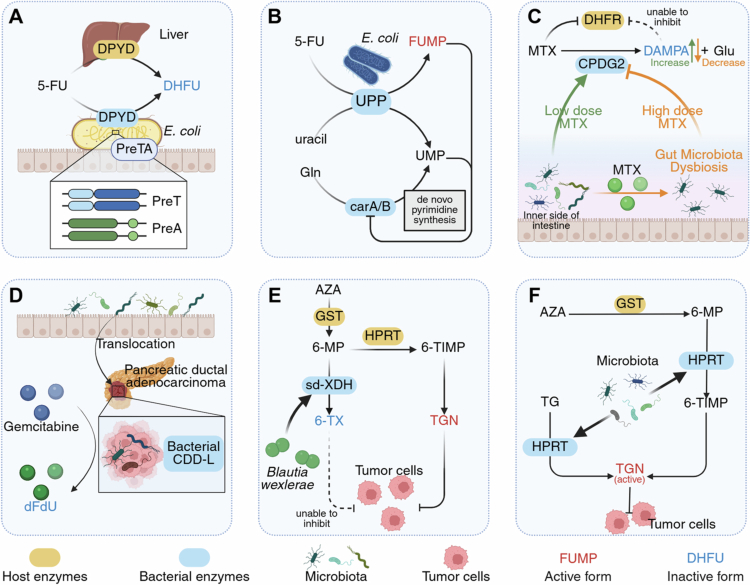 Figure 2
Figure 2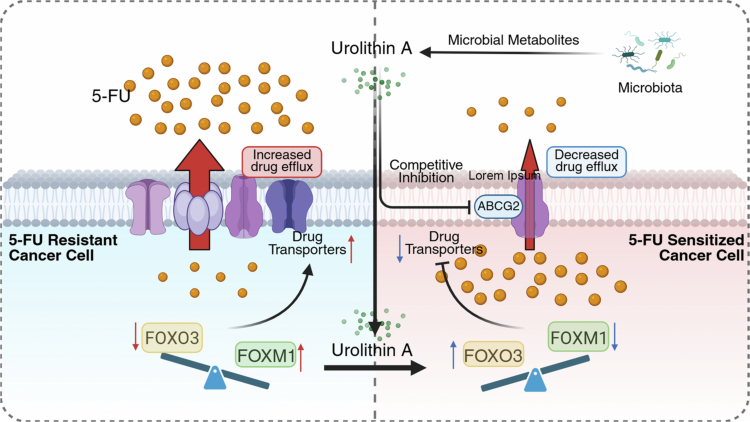 Figure 3
Figure 3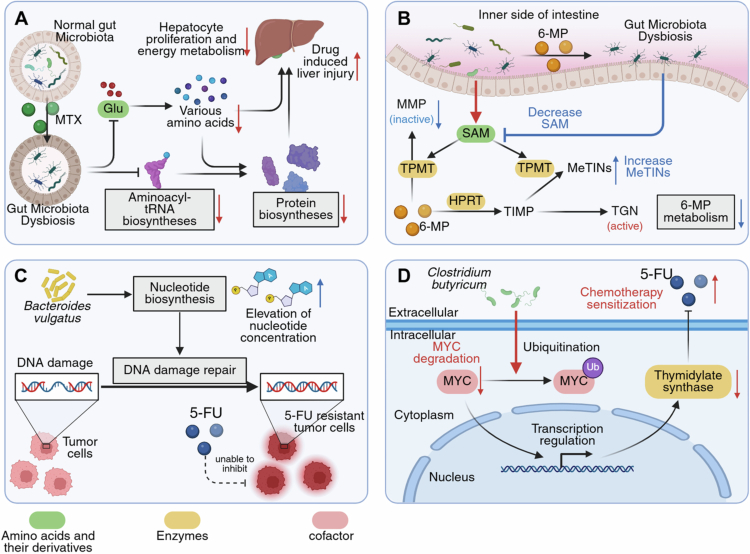 Figure 4
Figure 4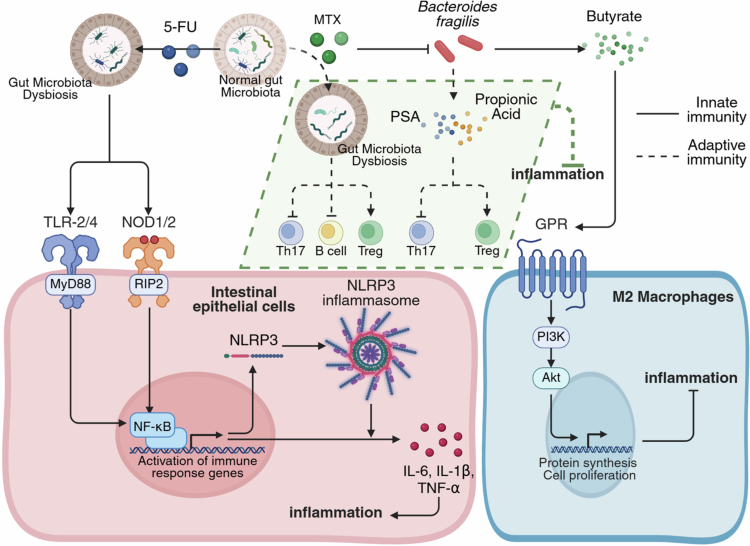 Figure 5
Figure 5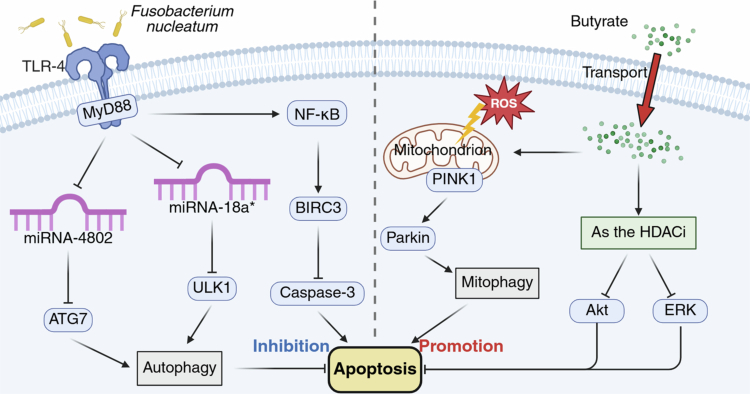 Figure 6
Figure 6| Chemotherapy | Microbiota feature | Effect | Reference | |
|---|---|---|---|---|
| 5-fluorouracil | Population | A reduction in | [ | |
| F/B ratio↓ | It stands for the imbalance in the ratio of | [ | ||
| Phylum | A decrease in | [ | ||
| Changes remain controversial, with factors such as animal/disease model type, dietary composition, drug dosage, treatment duration, and environmental differences potentially contributing to the discrepancies. | [ | |||
| [ | ||||
| An increase in | [ | |||
| Its increase is generally believed to promote intestinal inflammation. | [ | |||
| Genus | Changes in | [ | ||
| [ | ||||
| A decrease in | [ | |||
| Similar to | [ | |||
| Methotrexate | Population | At low doses, Methotrexate increases | [ | |
| [ | ||||
| F/B ratio↑* | At low doses, Methotrexate increases F/B ratio, while at high doses, it decreases F/B ratio. | [ | ||
| F/B ratio↓* | [ | |||
| Methotrexate | Phylum | At low doses, Methotrexate increases the abundance of | [ | |
| [ | ||||
| At low doses, Methotrexate decreases the abundance of | [ | |||
| [ | ||||
| Family | A decrease in | [ | ||
| Genus | It’s associated with the proliferation of pathogenic bacteria and the disruption of gastrointestinal tract health. | [ | ||
| A reduction in | [ | |||
| Species | A decrease in | [ | ||
| Gemcitabine | Population | Unknown. | [ | |
| Phylum | It downregulates the production of short-chain fatty acids and influences dietary energy absorption. | [ | ||
| Unknown. | [ | |||
| It leads to the rise of intestinal pathogens, such as | [ | |||
| Its increase is generally believed to promote intestinal inflammation. | [ | |||
| The decrease in | [ | |||
| Species | It has a dual effect, improving metabolism, restoring intestinal barrier function, and alleviating inflammation, while also degrading mucins and exacerbating intestinal inflammation. | [ | ||
| Capecitabine | Phylum | It downregulates the production of short-chain fatty acids and influences dietary energy absorption. | [ | |
| Unknown. | [ | |||
| It leads to the rise of intestinal pathogens, such as | [ | |||
| Genus | It leads to a weakened gut protective function and increased susceptibility to pathogenic bacteria. | [ | ||
| Mercaptopurine | Population | It weakens the protective effect of beneficial bacteria, allowing pathogenic species to colonize. | [ | |
| Phylum | It downregulates the production of short-chain fatty acids and influences dietary energy absorption. | [ | ||
| It leads to the rise of intestinal pathogens, such as | [ | |||
| Genus | Unknown. | [ | ||
| A reduction in | [ | |||
| Species | It has a dual effect, improving metabolism, restoring intestinal barrier function, and alleviating inflammation, while also degrading mucins and exacerbating intestinal inflammation. | [ | ||
| Tioguanine | Phylum | Unknown. | [ | |
|
| Unknown. | [ | ||
| Chemotherapy | Model | Response | Gut microbiota | Microbial gene functions | Reference |
|---|---|---|---|---|---|
| FOLFOX | Low-set rectal cancer patients | GR | [ | ||
| PR | |||||
| nCRT* | LARC patients | GR | [ | ||
| PR | |||||
| MTX | new-onset RA patients | GR | [ | ||
| MTX | new-onset RA patients | PR | [ | ||
| MTX | Psoriasis patients | GR | [ | ||
| PR | |||||
| MTX | RA patients | GR | Glycan biosynthesis and metabolism | [ | |
| PR | Vitamins biosynthesis and peptidoglycan biosynthesis | ||||
| MTX | RA patients | GR | Biosynthesis of several kinds of amino acids | [ | |
| PR | Pyruvate fermentation to acetate and lactate |
- —National Natural Science Foundation of China
- —Key Clinical Projects of Peking University Third Hospital
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Clostridium difficile and Clostridium perfringens research · Chemical Reactions and Isotopes
Introduction
Antimetabolite drugs are widely used chemotherapeutic agents that mimic the molecular structures of substances involved in nucleic acid synthesis, inhibiting cell division and proliferation. Antimetabolite drugs can be classified into five categories, based on the different target enzymes they inhibit: 1 thymidylate synthase (TS) inhibitors, such as 5-fluorouracil (5-FU);2 dihydrofolate reductase (DHFR) inhibitors, such as methotrexate (MTX);3 purine nucleotide interconversion inhibitors, such as mercaptopurine (6-MP);4 ribonucleotide reductase inhibitors, such as hydroxyurea; and 5 deoxyribonucleic acid (DNA) polymerase inhibitors, such as cytarabine. Owing to their broad cytotoxic activity, antimetabolite drugs are used to treat various malignancies (including colorectal cancer [CRC], breast cancer, leukemia, choriocarcinoma, and osteosarcoma) and inflammatory conditions (such as inflammatory bowel disease [IBD] and rheumatoid arthritis [RA]).1^,^2 This results in diverse adverse effects, including gastrointestinal, hepatic, pulmonary, neurological, and cardiotoxic effects.3^,^4 Persistent clinical challenges include treatment-induced gut microbiota disruptions. This disruption affects drug efficacy and toxicity, making it difficult to reliably predict patient responses. Therefore, it is essential to understand the mechanisms underlying these phenomena.
Recent advances in next-generation sequencing (NGS) technologies have enhanced our understanding of the gut microbiota. Notably, the critical influence of the gut microbiota on the efficacy of chemotherapy agents has been highlighted.5^,^6 This has led to the emergence of the field of “pharmacomicrobiomics” to describe their interactions.7^,^8 Antimetabolite drugs interfere with nucleic acid synthesis, consequently altering the composition and function of the host microbiota. This altered microbial state influences therapeutic outcomes through diverse mechanisms, highlighting the bidirectional relationship between microbiota and drug activity. Furthermore, the microbiota plays critical roles in the pharmacokinetics and pharmacodynamics of antimetabolite drugs, profoundly influencing their efficacy and side effects.9 Consequently, predicting the efficacy of antimetabolite drugs via microbiota profiling or enhancing drug outcomes by modulating microbial communities has become an increasingly active area of research.
Although most studies focus on the relationship between individual drugs and gut microbiota, comprehensive reviews systematically elucidating the complex interactions between antimetabolite drugs and the gut microbiota are lacking. In this review, we focus on several commonly used antimetabolite drugs, including 5-FU, MTX, gemcitabine, capecitabine, 6-MP, and thioguanine (TG). Their structures are shown in Figure 1. This review summarizes the effects of antimetabolite drugs on gut microbiota composition and elucidates their underlying mechanisms. We explore how the microbiota and its metabolites influence drug efficacy, highlight potential microbial biomarkers for predicting treatment responses, and discuss recent strategies targeting microbiota modulation to enhance therapeutic outcomes.
Structures of the antimetabolite drugs.
Impact of antimetabolite drugs on the gut microbiota
Antimetabolites alter the gut microbiota composition and metabolic activity (Table 1 shows the impact of antimetabolite drugs on the gut microbiota). Although drug-specific effects vary, similar trends have been reported. These include reduced alpha-diversity (α-diversity, to measure species richness and evenness), a decreased Firmicutes/Bacteroidetes (F/B) ratio (indicative of dysbiosis, as these phyla comprise approximately 90% of gut bacteria),10 depletion of beneficial taxa (e.g., Lactobacillus), and enrichment of potentially pathogenic/proinflammatory taxa (e.g., Proteobacteria). These microbial shifts may contribute to adverse effects, such as mucositis and gastrointestinal toxicity. However, most studies focus on certain drugs, such as 5-FU, necessitating further investigation across a wider range of antimetabolites.
5-Fluorouracil
2.1.
The uracil analog 5-FU exerts anticancer effects by inhibiting TS and incorporating its metabolites into ribonucleic acid (RNA) and DNA. 5-FU is primarily used in the treatment of various malignancies, including CRC, head and neck, breast, gastric, and skin cancers.61 Major adverse effects include cardiotoxicity, gastrointestinal toxicity, and myelosuppression.
Several studies have shown that 5-FU treatment reduces gut microbial α-diversity 11‐20 and the F/B ratio.13,16,21‐24 At the phylum level, most studies report decreased Firmicutes abundance and increased Proteobacteria and Verrucomicrobia abundance. However, the reported changes in Bacteroidetes abundance remain inconsistent. At the genus level, a consistent finding is decreased Lactobacillus abundance. The specific details of these microbial shifts are summarized in Table 1.
Alpha-diversity (α-diversity) measures within-sample species richness and evenness.62 It is a crucial determinant of gut microbiome health and influences the therapeutic responses. The intestinal microbiota protects the host by competitively excluding pathogens through a mechanism known as colonization resistance.63 A reduction in α-diversity weakens this protective barrier, allowing pathogen overgrowth, lipopolysaccharide (LPS) release, host inflammation,64 and impaired drug efficacy, as demonstrated in 5-FU–treated CRC mouse models.17 Another important indicator is the F/B ratio, reflecting the relative abundance of two predominant gut phyla and correlates with microbial homeostasis. Associations between the F/B ratio and age,65 IBD,66 diabetes,67 and obesity68 underscore its relevance to the host immune and metabolic status.
Phylum-level analyzes reveal distinct microbial shifts. Firmicutes, gram-positive bacteria vital for intestinal homeostasis and short-chain fatty acid (SCFAs) production,10 typically decrease, potentially exacerbating 5-FU–induced mucositis.16 Conversely, the abundance of gram-negative phyla, such as Proteobacteria and Verrucomicrobia, often increases. Elevated levels of Proteobacteria, which are normally a minor component69 but contain pathogens, indicate dysbiosis and contribute to the development of intestinal disease.70 An increased Verrucomicrobia abundance is associated with intestinal inflammation.25 Findings regarding Bacteroidetes, gram-negative polysaccharide degraders, remain contradictory across studies and are likely confounded by variations in experimental conditions, such as the model, diet, or treatment protocol.
Similar to the observations for Bacteroidetes, changes in the genus Bacteroides are inconsistent across studies, potentially reflecting its high variability among individuals.69 Another key genus is Lactobacillus (phylum Firmicutes), which ferments carbohydrate into lactic acid. These common residents of the human oral and gastrointestinal tracts are widely used as probiotics and exert crucial protective functions. A decline in Lactobacillus population is frequently associated with the overgrowth of pathogenic bacteria.
In addition to compositional changes, 5-FU significantly affects gut microbial function and gene expression. It alters genomic mutation profiles and markedly reduces G:C to T:A transversions. Evolutionary pressure can be assessed via the dN/dS ratio, which compares non-synonymous (dN) to synonymous (dS) substitution rates. This ratio appeared to stabilize (dN/dS = 80.1%) in most of the Kyoto Encyclopedia of Genes and Genomes (KEGG) functional modules.36 This widespread stability likely reflects purifying selection (dN/dS < 1), which preserves the core microbial functions under 5-FU treatment. This contrasts with neutral evolution (dN/dS ≈ 1, mutations are only affected by random genetic drift) and positive selection (dN/dS > 1, beneficial mutations are retained).71 Nevertheless, certain gene sets involved in cell proliferation and protein secretion showed elevated dN/dS ratios. This implies positive selection for adaptive strategies, such as enhanced proliferation or specific secretion, to survive 5-FU exposure. Additionally, 5-FU enriches microbial genes are related to the nicotinamide adenine dinucleotide (NAD) salvage pathway in patients. Activation of this pathway suppresses cancer cell autophagy and enhances survival. This finding suggests a possible mechanism by which the gut microbiota modulates therapeutic outcomes.
Methotrexate
2.2.
MTX is an anti-folate immunosuppressant, with folate being its primary target.72 MTX inhibits folate metabolism by competitively inhibiting DHFR, thereby blocking the de novo synthesis of purines and pyrimidines. MTX exerts dose-dependent effects.72 with low-dose MTX used for treating autoimmune diseases such as psoriasis, IBD, and RA73 and high-dose MTX used for cancers such as leukemia, lymphoma, and osteosarcoma.74 Common adverse effects of MTX include gastrointestinal, bone marrow, cardiac, neurologic, renal, and hepatic toxicities.3^,^4
MTX also exerts a dose-dependent effect on the gut microbiota (Table 1). Low doses generally increase α-diversity and the F/B ratio, illustrated by improved diversity in patients with RA40 and increased F/B ratio in gnotobiotic mice.47 On the other hand, high doses reduce diversity, induce inverse phylum shifts, and cause mucositis in healthy rats.44
Furthermore, MTX altered specific gut microbial taxa. Reported effects include reduced abundances of the family Ruminococcaceae, genera Lactobacillus and Prevotella, and the species Bacteroides fragilis (Table 1).
Such changes have functional implications, as the family Ruminococcaceae (phylum Firmicutes), comprising gram-positive bacteria, degrades resistant starch into SCFAs 75 and its depletion may impair gut degradation capabilities. At the genus level, Prevotella (phylum Bacteroidetes), a gram-negative bacterium, is enriched in patients with RA and associated with intestinal inflammation.76^,^77 Reduced Prevotella levels following MTX treatment may signify increased RA inflammation. Finally, the gram-negative species Bacteroides fragilis (phylum Bacteroidetes) plays a key role in immune modulation and suppression of inflammation;78 thus, its reduction might exacerbate inflammation.
MTX alters gut microbiota composition by targeting folate metabolism. Approximately 86% of the human gut microbial species are folate auxotrophs depending on external sources such as diet or cross-feeding, whereas only a minority (approximately 13%) are capable of de novo synthesis.79 This widespread dependence indicates that MTX-induced inhibition of folate metabolism leads to a broad suppression of the microbiota. For instance, the growth Prevotella, which requires external folate,80 is consequently stunted by MTX.81 Studies have demonstrated that folate-deficient diets decrease both α- and β-diversity in the fecal microbiota of healthy humans.82 This corroborates the role of folate inhibition in MTX-induced microbiota alterations.
Gemcitabine and capecitabine
2.3.
Gemcitabine is a deoxycytidine nucleoside analog and prodrug. After entering the human body, it is activated by deoxycytidine kinase to form gemcitabine di- and triphosphates. Gemcitabine diphosphate subsequently inhibits ribonucleotide reductase and gemcitabine triphosphate competes with deoxycytidine triphosphate for incorporation into DNA, ultimately inhibiting DNA synthesis and inducing apoptosis. Gemcitabine is widely used to treat ovarian, bladder, non-small cell lung, pancreatic, and breast cancer.83 Although gemcitabine can cause myelosuppression and gastrointestinal and pulmonary toxicity, its toxicity is generally milder than that of other pyrimidine antagonists.84 The primary challenges associated with gemcitabine include enzymatic deamination, rapid systemic clearance, and resistance.85
Gemcitabine increases α-diversity, reduces Firmicutes, Bacteroidetes, and Actinobacteria and increases Proteobacteria, Verrucomicrobia, and Akkermansia muciniphila. The specific microbial changes are listed in (Table 1).
Examining specific taxa provides further information regarding their functional impact. The phylum Actinobacteria, a group of gram-positive microbes found widely in nature, is a crucial source of metabolites, including most antibiotics and bioactive substances utilized by humans.86 Consequently, a reduction in Actinobacteria could signify a loss of beneficial genera, such as Bifidobacterium, thereby potentially compromising gut protection and increasing vulnerability to pathogens. A. muciniphila (gram-negative bacterium belonging to the phylum Verrucomicrobia) exhibits context-dependent functions. It promotes the host health by enhancing metabolism, reinforcing the gut barrier, and reducing inflammation.87 Conversely, it can degrade mucin, potentially worsening intestinal inflammation, particularly in malnourished hosts.88 Despite these potential microbial interactions, studies directly assessing the effects of gemcitabine on the human gut microbiota remain limited.
Capecitabine, an oral prodrug of 5-FU, is converted into 5-FU by thymidine phosphorylase, thereby exerting its antitumor effects.89 It has shown varying degrees of efficacy and acceptable tolerability in several cancers, including prostate, renal cell, ovarian, pancreatic, metastatic breast, and CRC cancers.90 Common toxicities of capecitabine include gastrointestinal and cardiovascular toxicities, as well as hand-foot syndrome.91
The effects of capecitabine on the gut microbiota included a decrease in Firmicutes at the phylum level, an increase in Bacteroidetes and Proteobacteria, and an increase in Bifidobacterium at the genus level. However, some studies have reported no significant effect of capecitabine on the gut microbiota,92 warranting further research (Table 1).
Bifidobacterium, a gram-negative bacterium from the Actinobacteria phylum, is a probiotic that can inhibit pathogenic bacteria and improve gut barrier function. It plays a regulatory role in various intestinal diseases including allergies, IBD, and cancer.93^,^94 Increased Bifidobacterium levels are beneficial for gut health.
Mercaptopurine and thioguanine
2.4.
The purine analogs 6-mercaptopurine (6-MP) and TG are important antimetabolites. 6-MP, often administered via its prodrug azathioprine (AZA) for potentially enhanced bioavailability,95 inhibits purine nucleotide biosynthesis and subsequent DNA synthesis. TG, a guanine analog, requires activation by hypoxanthine-guanine phosphoribosyl transferase to form cytotoxic nucleotides that are incorporated into and damage DNA, similar to the downstream effects of 6-MP.96 Both agents are used to treat acute lymphoblastic leukemia,97 and 6-MP is used to treat lymphoma and RA. Common primary toxicities include bone marrow suppression, gastrointestinal disturbances, and hepatic and renal dysfunction.98
6-MP reduces α-diversity, Firmicutes and Prevotella, and increases Proteobacteria, Bacteroides and A. muciniphila 59. The effect of TG on the gut microbiota is mainly at the phylum level, increasing Firmicutes and decreasing Bacteroidetes 60. However, research on these drugs is currently limited.
Influence of the gut microbiota and its metabolites on antimetabolite drug efficacy and toxicity
Emerging evidence highlights the influence of the gut microbiota on antimetabolite drug efficacy and toxicity. Although the general TIMER framework (Translocation, Immunomodulation, Metabolism, Enzymatic degradation, and reduced diversity/ecological variation) proposed by Alexander et al.99 describes chemotherapy-microbiota interactions, it may not fully capture the specificity of antimetabolites targeting nucleic acid biosynthesis. Therefore, we propose a framework that encompasses absorption, metabolism, distribution, excretion, endogenous metabolite production, immune modulation, and apoptotic pathway modulation, to delineate the specific mechanisms underlying gut microbes regulation of antimetabolite outcomes.
Microbe-induced changes to drug pharmacokinetics
3.1.
Microbe-induced drug absorption
3.1.1.
The gut microbiota regulates intestinal permeability by modulating the integrity of tight junctions and thickness of the intestinal mucus layer, ultimately influencing the absorption of pharmacological agents.
Antimetabolite drugs can cause gut microbiota dysbiosis exacerbating damage to tight junction proteins among intestinal epithelial cells and disrupting the cell surface mucus layer, thereby increasing intestinal epithelial permeability.26 In contrast, probiotic interventions strengthen intercellular tight junctions, increase mucus layer thickness, and decrease permeability.100 The state of intestinal tight junctions and the mucus layer profoundly impacts drug diffusion, epithelial absorption, and overall bioavailability. Studies have confirmed that targeted modulation of tight junctions can facilitate paracellular drug transport.101 In contrast, a thickened mucus layer inhibits effective drug delivery.102
However, few studies have specifically investigated the effects of these structural changes on drug absorption, as most research has focused on the status of tight junctions and the mucus layer as indicators of chemotherapy-induced intestinal barrier disruption and mucositis.14 Thus, the influence of the gut microbiota on antimetabolite drug absorption remains incompletely understood.
Microbe-induced drug metabolism
3.1.2.
The gut microbiota contains millions of protein-coding genes capable of metabolizing diverse nutrients and altering drug pharmacokinetics.7 Increasing evidence indicates that bacteria-derived enzymes can influence the response and toxicity of antimetabolite drugs.99
The preTA operon in the gut microbiota encodes dihydropyrimidine dehydrogenase (DPYD), that reduces the bioavailability and chemotherapeutic efficacy of 5-FU by metabolizing it to the inactive metabolite dihydrofluorouracil (DHFU) (Figure 2A). In the mammalian liver, DPYD metabolizes 5-FU to inactive DHFU. Recent findings have shown that the preTA operon in Escherichia coli encodes DPYD and metabolizes 5-FU, thereby reducing the bioavailability and efficacy of oral 5-FU treatment in mice.103 Moreover, cell-free supernatants from preTA-overexpressing E. coli significantly reduce the inhibitory effect of 5-FU on tumor cells.103 The preTA operon is commonly found in the human gut, particularly in Proteobacteria and Firmicutes 103. The genome of Anaerostipes hadrus also harbors the preTA operon and can metabolize 5-FU into DHFU; however, it also contains hydA, which encodes bacterial dihydropyrimidinase, enabling the metabolism of DHFU into α-fluoro-β-ureidopropionic acid.104 However, the hydA gene in E. coli is located further from the preTA operon; therefore, 5-FU metabolism is halted at DHFU.103^,^104
Mechanisms by which the gut microbiota alters drug metabolism and affects the activity of antimetabolite drugs. A The preTA operon in E. coli can express enzymes similar to DPYD in the host liver and degrade 5-FU to the inactive metabolite DHFU. B The UPP enzyme in E. coli metabolizes 5-FU to FUMP, which blocks the de novo pyrimidine synthesis pathway and inhibits tumor growth. C At low doses, MTX is metabolized by CPDG2 in the gut microbiota to the inactive metabolite DAMPA, but at higher doses, MTX-induced dysbiosis downregulates CPDG2, delaying DAMPA generation. D A CDD-L enzyme present in the gut microbiota translocated to pancreatic tumors can degrade gemcitabine to the inactive metabolite dFdU. E Blautia wexlerae-derived sd-XDH degrades AZA or 6-MP to the inactive metabolite 6-TX. F The gut microbiota expresses enzymes similar to those of the host HPRT, facilitating the conversion of 6-MP, AZA, and TG into the active metabolite TGN. 5-FU, 5-fluorouracil; 6-MP, mercaptopurine; 6-TIMP, 6-thioinosine monophosphate; 6-TX, 6-thioxanthine; AZA, azathioprine; carA/B, carbamoyl phosphate synthetase; CDD, cytidine deaminase; CPDG2, carboxypeptidase glutamate 2; DAMPA, 2, 4-diamino-N-10-methylpteroic acid; DHFR, dihydrofolate reductase; DHFU, dihydrofluorouracil; dFdU, 2',2'-difluorodeoxyuridine; DPYD, dihydropyrimidine dehydrogenase; FUMP, 5-fluorouridine monophosphate; Gln, glutamine; Glu, glutamic acid; GST, glutathione S-transferase; HPRT, hypoxanthine phosphoribosyltransferase; MTX, methotrexate; sd-XDH, selenium-dependent xanthine dehydrogenase; TGN, 6-thioguanine nucleotide; UMP, uridine monophosphate; upp, uracil phosphoribosyltransferase.
A recent prospective cohort study used metagenomic sequencing to corroborate the interaction between preTA and oral fluoropyrimidines, including the 5-FU prodrug capecitabine,105 revealing that chemotherapy enriches pyrimidine metabolism genes (such as preTA and upp) in the human gut microbiome—explaining why gastrointestinal toxicity often appears early in treatment and later subsides; and that oral fluoropyrimidine toxicity is microbiome-dependent, with bacterial preTA mitigating adverse effects. Furthermore, a predictive model based on baseline preTA levels demonstrated high accuracy in assessing patient treatment toxicity. Collectively, these studies indicate that bacterial preTA has significant clinical potential as a crucial biomarker and interventional target for regulating drug efficacy and toxicity.
Uracil phosphoribosyltransferase (UPP) in the gut microbiota converts 5-FU to 5-fluorouridine monophosphate (FUMP), thereby increasing chemotherapy efficacy (Figure 2B). A study using Caenorhabditis elegans suggested that 5-FU and its prodrug derivative, 5-fluoro-2′-deoxyuridine (FUDR), act via the regulation of ribonucleotide rather than DNA metabolism.106 Both 5-FU and FUDR are metabolized to FUMP in E. coli via the pyrimidine salvage pathway, which is encoded by an upregulated gene.106 Uridine monophosphate (UMP) blocks de novo pyrimidine synthesis in both bacteria and hosts by inhibiting the carA/B enzymes. It has been hypothesized that FUMP, a structural analog of UMP, may exert a similar effect; however, this has not been experimentally validated. By converting 5-FU and FUDR into FUMP, microbial enzymes may block de novo pyrimidine synthesis, thus inhibiting tumor growth.106
Carboxypeptidase glutamate 2 (CPDG2) in the gut microbiota converts MTX into inactive 2, 4-diamino-N-10-methylpteroic acid (DAMPA), thereby reducing MTX efficacy and mitigating its toxicity (Figure 2C). CPDG2 inactivates MTX and its metabolite 7-OH-MTX by cleaving the terminal glutamate residues, yielding DAMPA and 7-OH-DAMPA, respectively.41^,^107 The essential role of the microbiota in this conversion is confirmed by the absence of DAMPA in germ-free or antibiotic-treated mice.108 p-aminobenzoyl-glutamate hydrolase found in E. coli is also capable of this reaction.109 As DAMPA does not inhibit DHFR, this microbial activity reduces the effects of MTX. Clinically, CPDG2 has been approved as an antidote for delayed MTX clearance.110
Importantly, MTX-induced microbiota alterations downregulate CPDG2, potentially resulting in delayed MTX detoxification (Figure 2C). At low MTX doses, 7-OH-MTX is excreted within 12 h, whereas DAMPA is excreted between 12 and 48 h. However, at higher MTX doses, the excretion of MTX and DAMPA is delayed to 48 h.41 High-dose MTX-induced alterations in the gut microbiota may reduce CPDG2 activity, thereby delaying MTX detoxification into DAMPA.41 Specifically, high-dose MTX may decrease the abundance of bacterial families, such as Prevotellaceae, Anaeroplasmataceae, Ruminococcaceae, and Lactobacillaceae, that are positively correlated with DAMPA excretion and likely contribute to CPDG2 production. These alterations could impair MTX clearance by decreasing overall CPDG2 activity.41
Folylpolyglutamate synthase (FPGS) in the gut microbiota can convert MTX into MTX-PG, although its effect on the therapeutic outcomes of MTX may be minimal. Certain gut bacteria can add glutamate to MTX via FPGS to generate MTX-PG,111 that displays a greater affinity for its target proteins compared to MTX. However, it has a lower affinity for the reduced folate transporter and proton-coupled folate transporter, which complicates its transport. Therefore, MTX-PGs produced by the gut microbiota are unlikely to significantly affect the therapeutic efficacy of MTX.112
Cytidine deaminase (CDD) in intratumoral microorganisms found in pancreatic ductal adenocarcinoma (PDAC) converts gemcitabine into an inactive metabolite, thereby reducing its efficacy (Figure 2D). Geller et al. reported that the long isoform of bacterial CDD (CDD-L) from Mycoplasma hyorhinis in human dermal fibroblasts can convert gemcitabine into the inactive 2′,2′-difluorodeoxyuridine, thus mediating gemcitabine resistance.113 Similarly, CDD from Enterococcus faecium mediates gemcitabine resistance in gallbladder cancer.114 Consistently, antibiotic treatment or treatment with CDD-deficient E. coli significantly improved the antitumor response to gemcitabine in mice. Geller et al. further investigated intratumoral bacteria in human PDAC,113 revealing that the proportion of tumors containing bacteria (76%) was significantly higher than that in the normal pancreas (15%) and the most common species detected was Gammaproteobacteria. The authors hypothesized that these bacteria migrate retrogradely from the duodenum to the pancreas. Pushalkar et al. further confirmed this hypothesis using bacterial translocation experiments, which revealed that fluorescently-labeled Enterococcus faecalis migrated into the pancreas of mice,115 suggesting direct influence of gut bacteria on the pancreatic microenvironment. The gut and pancreatic microbiomes of patients with PDAC exhibited similar community profiles, with enrichment of Proteobacteria. Recent studies have suggested that fecal microbiota transplantation (FMT) can modulate the characteristic microbiome enrichment observed in patients with PDAC.116 This modulation creates a favorable tumor microenvironment and potentially improves patient prognosis. Similar to the gut microbiota, a recent study demonstrated that oral bacteria also contribute to gemcitabine resistance in pancreatic cancer via CDD.117 Oral bacteria may invade the pancreatic tissue through hematogenous spread or gastrointestinal translocation, leading to gemcitabine degradation. This study emphasized that the enzymatic function of CDD is independent of isoform length, contrasting with an earlier study that demonstrated that the long isoform is fully functional, whereas the short isoform retains only partial functionality.113
Gut microbial selenium-dependent xanthine dehydrogenase (sd-XDH) inactivates 6-MP, the active metabolite of AZA, by converting it to 6-thioxanthine (6-TX), thereby reducing the therapeutic effectiveness of AZA in IBD (Figure 2E). Specifically, Blautia wexlerae-encoded sd-XDH converts 6-MP to 6-TX, which decreases 6-thioinosine-monophosphate (6-TIMP), a precursor of the active drug form, thioguanine nucleotide (TGN), ultimately, contributing to AZA treatment failure in IBD.118
Gut microbial hypoxanthine phosphoribosyltransferase (HPRT) converts TG into its active form, TGN (Figure 2F). Recent evidence suggests that TG exerts therapeutic effects in IBD through bacterial metabolism, independent of the adaptive immune system. Although HPRT is considered critical for this conversion, TG has shown efficacy in HPRT-deficient mice without systemic immunosuppression.60 In vitro studies have demonstrated that E. coli, E. faecalis, and Bacteroides thetaiotaomicron can produce TGN from TG, implicating the involvement of bacterial HPRT. Strategies to enhance TG efficacy may involve modulating the intestinal microbiota or optimizing drug delivery to increase the colonic residence time.119 Notably, Bacillus subtilis, which also encodes HPRT, increases 6-TIMP levels, thereby rescuing AZA treatment failure induced by Blautia wexlerae in IBD.118
Microbe-induced drug distribution
3.1.3.
Gut microbiota and its metabolites can regulate the distribution of drugs by modulating drug transporters on tumor cell surfaces, thereby affecting the efficacy and toxicity of antimetabolite drugs.
Urolithins downregulate various drug transporters, reduce the efflux of 5-FU, and enhance drug efficacy. Urolithin is a gut microbiota metabolite derived from the dietary polyphenols ellagitannins and ellagic acid, divided into urolithin A (UroA) and urolithin B (UroB).120 Urolithins, especially UroA, demonstrate broad antitumor activities.121 UroA and its structural analog, UAS03, downregulate the expression of drug transporters (MDR1, BCRP, MRP2, and MRP7), thereby reducing the efflux of 5-FU and sensitizing cancer cells.122 Specifically, forkhead box (FOX) proteins play crucial roles, with FOXM1 and FOXO3 acting as upstream regulators of drug transporters. FOXM1 enhances the expression of the drug transporter ABCC10 and mediates 5-FU resistance. Conversely, FOXO3 directly inhibits FOXM1 and antagonizes FOXM1 function by competing for the same target genes.123 Therefore, elevated FOXM1 and decreased FOXO3 expression are associated with resistance to cancer therapy. UroA/UAS03 reduces the expression of drug transporters by regulating the FOXO3-FOXM1 axis, thus sensitizing CRC cells to 5-FU (Figure 3).
Mechanisms by which urolithin A downregulates drug transporters. The metabolite urolithin A produced by the gut microbiota regulates the FOXO3-FOXM1 axis, leading to reduced expression of drug transporters on the tumor surface and decreased efflux of 5-FU. Additionally, urolithin A can competitively inhibit the drug transporter ABCG2, further reducing the efflux of 5-FU. 5-FU, 5-fluorouracil; FOXM1, forkhead box M1; FOXO3, forkhead box O3.
Additionally, ATP-binding cassette (ABC) transporters are crucial for 5-FU transport during chemotherapy.124 Their subfamilies B (e.g., ABCB1), C (e.g., ABCC1/2/3), and G (e.g., ABCG2) mediate drug efflux. UroA reverses 5-FU resistance by inhibiting ABCG2125 (Figure 3). In contrast, butyrate—a gut microbiota-derived SCFA and histone deacetylase inhibitor with antitumor activity126—is potentially associated with 5-FU resistance via ABCG2. Chronic butyrate exposure downregulates its transporter, sodium-coupled monocarboxylate transporter 1 (SMCT1), causing butyrate resistance in CRC cells.127 Butyrate-resistant CRC cells (HCT-116/BR) show cross-resistance to 5-FU, possibly due to their high ABCG2 expression.128^,^129 Notably, Lactobacillus plantarum-derived metabolites can overcome this dual resistance by upregulating SMCT1.127
Microbe-induced drug excretion
3.1.4.
The gut microbiota indirectly influences the excretion of antimetabolites, primarily by modulating the systemic burden of the active drug and its metabolites. For instance, gut microbes metabolize MTX into the non-toxic DAMPA via CPDG2. The enzymatic activity of microbial CPDG2 directly impacts the elimination rates of both 7-OH-MTX and DAMPA.41 Thus, microbial metabolic inactivation of antimetabolites is frequently and intimately tied to overall drug excretion.
Additionally, the gut microbiota can subject drugs to repeated metabolism through enterohepatic circulation, thereby affecting hepatic excretion. In contrast, its influence on renal excretion is more indirect, largely affecting the excretory load of the kidney based on the systemic concentrations of the parent compound and its derivatives.
Microbe-induced endogenous metabolite production
3.2.
Microbiota-derived endogenous metabolites—including amino acids, nucleotides, and regulatory factors—modulate host responses to antimetabolite drugs via multiple pathways.
Gut microbiota enhances MTX-induced liver injury by influencing aminoacyl-tRNA biosynthesis. Given the critical role of the liver in amino acid homeostasis and the association between disrupted amino acid metabolism and liver toxicity,130 modulation of aminoacyl-tRNA, which is essential for protein synthesis, is significant. Specifically, Ruminococcus and Lactobacillus promote aminoacyl-tRNA biosynthesis, whereas Collinsella and Streptococcus inhibit it. MTX perturbs these microbial populations, suppressing aminoacyl-tRNA biosynthesis and downregulating amino acids, such as glycine, L-aspartic acid, and L-valine, ultimately leading to reduced protein synthesis and drug-induced liver injury (DILI)48 (Figure 4A).
Mechanisms by which the gut microbiota regulates various endogenous metabolites and affects the efficacy of antimetabolite drugs. A MTX-induced dysbiosis inhibits the biosynthesis of aminoacyl-tRNA, downregulates the metabolism of various amino acids, such as glutamic acid, affects hepatocyte proliferation and energy metabolism, and mediates drug-induced liver injury. B 6-MP-induced dysbiosis downregulates SAM concentrations and inhibits TPMT activity, thereby affecting the metabolism of 6-MP. C Bacteroides vulgatus-mediated nucleotide biosynthesis promotes DNA damage repair in tumor cells, conferring resistance to 5-FU. D Clostridium butyricum promotes the ubiquitination and degradation of MYC, leading to the downregulation of thymidylate synthase. 5-FU, 5-fluorouracil; 6-MP, mercaptopurine; Glu, glutamic acid; HPRT, hypoxanthine phosphoribosyltransferase; MeTINs, methylthio-6⁃thioinosine phosphates; MMP, 6-methylmercaptopurine; MTX, methotrexate; SAM, S-adenosylmethionine; TGN, thioguanine nucleotide; TIMP, thioinosine monophosphate; TPMT, thiopurine S-methyltransferase; Ub, ubiquitin.
Furthermore, the gut microbiota contributes to liver damage by modulating glutamic acid metabolism. Glutamic acid is a multifunctional amino acid that functions as a neurotransmitter and participates in the metabolism of several other amino acids. Lactobacillus, Streptococcus, Enterococcus, and other bacteria are associated with glutamic acid metabolism. MTX reduces the abundance of gut microbes by downregulating glutamic acid and its associated amino acid metabolic pathways, impairing hepatocyte proliferation and energy metabolism, ultimately leading to DILI48 (Figure 4A).
Tryptophan (TRP) metabolism is another critical pathway regulated by gut microbiota. Research has shown that the probiotic Weissella paramesenteroides WpK4 alleviates 5-FU-induced intestinal mucositis131 by producing and catabolizing TRP and generating endogenous metabolites such as indole-3-acetic acid (IAA) and tryptamine. IAA acts as a ligand and activates the aryl hydrocarbon receptor (AhR), which translocates from the cytoplasm to the nucleus where it forms a heterodimeric complex with the AhR nuclear translocator. This complex initiates the transcription of specific target genes and plays a crucial role in local immune response. Prebiotic dried ginger essential oil also modulates TRP metabolism in the gut microbiota,132 generating metabolites including IAA, 5-hydroxyindole-3-acetic acid, and oxoadipic acid, thereby mitigating 5-FU-induced mucositis. This study proposed an IAA-based AhR/interleukin (IL)22/signal transducer and activator of transcription 3 (STAT3) signaling axis, where AhR activation by IAA induces the expression of downstream cytokines, such as IL-22 and IL-17,133 with IL-22 subsequently activating the STAT3 pathway. This pathway is essential for regulating and maintaining intestinal barrier integrity while suppressing apoptosis-related processes.134 In addition to IAA, other TRP metabolites influence chemotherapy efficacy. Under high-salt diet conditions, the gut bacterial TRP metabolism affects FOLFOX efficacy via immunomodulation.135 The TRP metabolites indole, skatole (SK), indole propionic acid (IPA) and indole-3-carboxaldehyde significantly increased IL-1β and TNF-α production in macrophages. In contrast, tryptophan, tryptamine, and indole-3-lactic acid significantly decreased them. Interestingly, although IAA promoted the efficacy of FOLFOX upon long-term exposure, it inhibited its efficacy during short-term treatment. This study further demonstrates that SK suppression and IPA enhancement potentiate the therapeutic effects of FOLFOX, although the underlying mechanisms remain unclear.
Gut microbiota modulates 6-MP metabolism via S-adenosylmethionine (SAM) production. SAM serves as a ubiquitous methyl donor that is critical for cellular methylation.136 Treatment with 6-MP induces gut dysbiosis and reduces SAM levels in the cecum and plasma, thereby impairing the stability and activity of thiopurine S-methyltransferase, an enzyme dependent on SAM for 6-MP methylation.59 Consequently, 6-MP metabolism shifts are characterized by decreased formation of the primary methylated metabolite 6-methylmercaptopurine and increased accumulation of secondary methylated products, methylthio-6-thioinosine phosphates59 (Figure 4B). Bacteria in the genus Alistipes are implicated in SAM dysregulation.59
The gut microbiota also promotes FOLFOX resistance by increasing SAM concentration.137 Desulfovibrio desulfuricans and its metabolites elevate serum SAM levels in the gut. As a crucial intracellular methyl donor, SAM interacts with methyltransferase-like 3 (METTL3), increasing the number of catalyzed RNA methylation modification reactions. Upregulation of METTL3 via the JAK2-STAT3 pathway leads to the expression of pro-oncogenic genes and confers FOLFOX resistance in CRC cells. METTL3 silencing can counteract this resistance, thereby restoring chemotherapeutic efficacy. However, the impact of SAM on tumors remains controversial. One perspective suggests that, at low concentrations, SAM upregulates METTL3 and promotes tumor progression,137 whereas at high concentrations, it induces cell death and autophagy in tumor cells.138 The effect of SAM is likely dominated by the former mechanism under the influence of gut microbiota, rather than exogenous supplementation.
Gut microbes can diminish the efficacy of 5-FU by increasing endogenous nucleotide production, which enhances DNA repair in tumors. For instance, Bacteroides vulgatus levels are significantly elevated in patients with locally advanced rectal cancer who respond poorly to neoadjuvant chemoradiotherapy.20 This bacterium promotes resistance by stimulating the de novo nucleotide biosynthesis pathway, thereby facilitating tumor DNA repair and protecting cancer cells from chemotherapy-induced damage20 (Figure 4C). Consistent with this mechanism, the administration of exogenous nucleosides or B. vulgatus in mouse colorectal tumor models significantly reduced the therapeutic effect of 5-FU.
Host ribonucleotide and deoxyribonucleotide metabolism, which is regulated by microbial communities, critically influences 5-FU efficacy.139 Studies on Caenorhabditis elegans show that bacteria affect 5-FU by participating in ribonucleotide metabolism, facilitating the activation of 5-FU (e.g., FUMP/FUTP) and by modulating the host deoxyribonucleotide pool, thereby altering downstream effects of 5-FU TS inhibition, including DNA damage.139 The role of ribonucleotide metabolism in 5-FU activation has been supported by multiple studies,106^,^139 albeit with different interpretations. Earlier work highlighted UPP-mediated FUMP production and pyrimidine synthesis inhibition,106 whereas Scott et al. emphasized the subsequent conversion to FUTP, leading to RNA damage and cell death.139 Regarding the deoxyribonucleotide pathway, Scott et al. proposed that bacterial modulation of host deoxyribonucleotide levels induces DNA damage and bacteria-dependent autophagy, thereby altering 5-FU response.139 In contrast, Bacteroides vulgatus promotes DNA repair via nucleotide biosynthesis.20 However, Scott et al. suggested that a bacterial deoxyribonucleotide imbalance could influence 5-FU efficacy independent of DNA repair.139 Collectively, these results suggest that bacterial deoxyribonucleotide metabolism potentially affects 5-FU outcomes through both autophagy and DNA damage/repair pathways.
The gut microbiota-derived secondary bile acid, deoxycholic acid (DCA) enhances the efficacy of FOLFOX in CRC.140 DCA mediates this enhancement by upregulating the AOX3/Cyp26b1 pathway through activation of Ugt1a6b, a key component of the enterohepatic circulation of bile acids. Ugt1a6b encodes the UDP-glucuronosyltransferase enzyme, which plays a pivotal role in bile acid enterohepatic circulation. The combination of DCA and FOLFOX increased the mRNA expression of Ugt1a6b and other genes associated with enterohepatic circulation. This promotes the glucuronidation of bile acids and improves enterohepatic cycling, thereby enhancing FOLFOX reabsorption and increasing its therapeutic efficacy. Further investigations are required to determine whether other bile acids exert similar effects.
Clostridium butyricum influences 5-FU efficacy by modulating regulatory factors. This bacterium enhances proteasome-mediated ubiquitination and subsequent degradation of the MYC oncogene, which in turn promotes the transcription of thymidylate synthase (TS). TS is a key target enzyme of 5-FU, and its overexpression often causes 5-FU resistance.141 Therefore, C. butyricum-mediated MYC degradation reduces TS expression, which sensitized tumors to 5-FU treatment142 (Figure 4D), thereby leveraging the known tumor-suppressive effect of MYC inactivation.143
Microbe-induced immune modulation
3.3.
Growing evidence highlights the capacity of the gut microbiota to modulate host immunity.144 In the context of antimetabolite chemotherapy, this microbial influence affects treatment efficacy by modulating tumor and inflammatory immunity, as well as treatment toxicity by altering inflammatory responses underlying side effects such as enteritis.145 The microbiota regulates both the innate and adaptive immune systems. Importantly, this immunomodulation is not straightforward potentiation or inhibition but a complex, bidirectional interaction often described as a “double-edged sword.”
Innate immunity
3.3.1.
The gut microbiota interacts with various pattern recognition receptors (PRRs) to regulate the innate immune response, including toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs).
TLRs are a family of PRRs that recognize conserved microbial structures (pathogen-associated molecular patterns) and host-derived danger signals (damage-associated molecular patterns).146 Continual interactions between gut microbes and the epithelium result in physiological low-grade inflammation. In contrast, chemotherapy-induced dysbiosis disrupts this homeostasis, often hyperactivating TLR pathways and promoting inflammatory damage.147 Canonical TLR signaling typically proceeds via the myeloid differentiation primary response gene 88 (MyD88)-dependent pathway, culminating in nuclear factor-κB (NF-κB) activation and proinflammatory cytokine secretion.148
The roles of TLR2 (which detects gram-positive bacteria) and TLR4 (which detects gram-negative bacteria) are well established in intestinal inflammation.149 5-FU-induced dysbiosis activates both TLR2/MyD88/NF-κB and TLR4/MyD88/NF-κB signaling pathways in rats, elevating inflammatory cytokines such as IL-1β, IL-6, and TNF-α18^,^26 (Figure 5). Specific bacterial taxa correlate with this signaling: certain gram-positive genera (Aerococcus, Blautia, Roseburia) are inversely associated with TLR4 activity, whereas gram-negative genera (Bacteroides, Parabacteroides, Parasutterella) show positive correlations and may exacerbate inflammation.21 Conversely, probiotic interventions such as Akkermansia supplementation can alleviate inflammation by downregulating TLR2 and inhibiting NF-κB signaling.150
Mechanisms by which the gut microbiota influences drug efficacy through immune regulation. In terms of innate immunity, the microbiota activates the TLR-2/4 and NOD1/2 receptors, promoting the expression of the NLRP3 inflammasome and various inflammatory cytokines. The metabolite butyrate also stimulates M2 macrophage proliferation through GPR receptors. In adaptive immunity, the microbiota and their metabolites regulate the proportions of various immune cells. 5-FU, 5-fluorouracil; GPR, G protein-coupled receptor; MTX, methotrexate; NLRP-3, NOD-like receptor thermal protein domain associated protein 3; NOD, nucleotide binding oligomerization domain; PSA, polysaccharide A; TLR, Toll-like receptor.
Microorganisms also influence immune responses via NLRs. Unlike TLRs, which detect extracellular or endosomal ligands, NLR proteins NOD1 and NOD2 function as cytosolic PRRs that recognize bacterial peptidoglycans within host cells.151 Upon ligand recognition, NOD1 and NOD2 oligomerize to activate receptor-interacting protein 2 (RIP2), which triggers downstream signaling cascades involving both NF-κB and mitogen-activated protein kinase (MAPK) pathways, ultimately driving the transcription of immune-related genes and exacerbating RA.152^,^153 5-FU-induced microbiota dysbiosis upregulates NOD1/2 expression, activating NOD/RIP2/NF-κB signaling and altering the secretion of inflammatory cytokines including IFN-γ, TNF-α, IL-1β, and IL-6,30^,^154 Figure 5.
NOD-like receptor thermal protein domain-associated protein 3 (NLRP-3), anotheran important member of the NLR family, also influences the immune response. NLRP-3 forms a multiprotein signaling complex (NLRP-3 inflammasome) under pathogenic stimulation.155 Once activated, it induces the production of proinflammatory cytokines such as IL-1β and IL-18, thereby causing inflammation and facilitating tissue and organ regeneration.156 NLRP-3 inflammasome is highly expressed in intestinal tissues of mice with 5-FU-induced mucositis, where sustained PRR stimulation by harmful bacteria activates the NLRP-3 inflammasome, thereby increasing TNF-α, IL-6, IL-1β, and IL-18 levels and exacerbating inflammation157 (Figure 5). Beneficial microbes such as A. muciniphila and its outer membrane protein Amuc_1100 reduce the activation of NLRP-3 inflammasomes, alleviating 5-FU-induced mucositis in mice.158
The gut microbiota also modulates innate immunity by influencing macrophage activation and polarization.159 For instance, B. fragilis promotes anti-inflammatory M2 macrophages, critical for regulating inflammatory diseases.160 Notably, B. fragilis abundance diminishes following MTX treatment in both preclinical models and patients with RA.3^,^49 MTX efficacy in collagen-induced arthritis models is dependent on B. fragilis, with its transplantation restoring MTX activity in microbiota-depleted mice.49 This bacterium appears to enhance the efficacy of MTX by increasing the M2 macrophage population49 (Figure 5), potentially via butyrate production. B. fragilis elevates butyrate levels, and butyrate alone can restore MTX efficacy in such models.49 Butyrate promotes M2 proliferation and dampens inflammation via G protein-coupled receptor mediated phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling.161 In contrast, colonization with Bacteroides wexlerae promotes inflammatory macrophages and reduces AZA efficacy in a mouse model of colitis,118 emphasizing species-specific effects on drug response.
Adaptive immunity
3.3.2.
Adaptive immunity, including both humoral and cell-mediated pathways, is also modulated by the intestinal microbiota. In the context of antimetabolite drug therapy, microbial regulation mainly affects cell-mediated immune responses.
The gut microbiota influences T-cell proliferation and differentiation, potentially enhancing the anti-inflammatory efficacy of MTX. MTX functions through multiple host mechanisms (including nucleotide synthesis inhibition, adenosine release, reactive oxygen species [ROS], and cytokines),162 and also targets microbial pathways, reducing immune activation.51^,^163 Transferring MTX-altered microbiota into germ-free mice reduces activated T cell populations, including T helper 17 (Th17) cells, suggesting that bacteria such as Prevotella copri and Collinsella aerofaciens mediate adaptive immune suppression after MTX treatment51 (Figure 5). Clinically, MTX combined with glucocorticoids helps restore the Th17/regulatory T cell (Treg) balance in patients with early RA by modulating the microbiota,42 thereby targeting Th17-driven inflammation in RA.164
B. fragilis promotes Treg proliferation and suppresses adaptive immunity via metabolites. Its immunoregulatory molecule, polysaccharide A, inhibits IL-17 production while inducing CD4 + T cell differentiation into IL-10-producing Foxp3 + Tregs, ameliorating colitis in animal models.165 B. fragilis also utilizes SCFAs for immunoregulation in conditions such as IBD.166 The YCH46 strain of B. fragilis produces propionic acid, which increases Tregs and decreases Th17 cells, thereby reducing inflammation 167 (Figure 5).
Parabacteroides distasonis also influences T cell responses. A study on high-dose MTX for primary central nervous system lymphoma revealed a correlation between gut P. distasonis abundance and prognosis.168 This bacterium may act via the gut-brain axis; its higher abundance is associated with increased CD8+ T cell infiltration in the cerebrospinal fluid, a marker of favorable outcomes. Further analysis indicated a positive correlation between P. distasonis abundance and plasma betaine-valerate levels. Thus, P. distasonis can slow disease progression by indirectly promoting CD8+ T cell infiltration into the cerebrospinal fluid and tumor tissue.
Microbe-induced apoptotic pathway modulation
3.4.
Antimetabolite drugs can induce apoptosis in tumor cells by inhibiting DNA biosynthesis and repair as well as through various apoptotic pathways. For example, 5-FU can activate the death receptor tumor necrosis factor receptor 1 by upregulating TNF-α, leading to the initiation of the extrinsic apoptotic pathway, which causes the activation of caspase-3 and caspase-8, culminating in cell apoptosis.23 Studies have demonstrated that gut microbiota can significantly alter the apoptosis-inducing therapeutic efficacy of antimetabolite drugs through multiple apoptotic pathways.
Fusobacterium nucleatum, a pathogenic bacterium, mediates 5-FU resistance by suppressing apoptotic pathways in colon cancer cells.169^,^170 F. nucleatum can specifically activate the TLR4 and MYD88 immune signaling pathways in tumor cells, inhibiting specific microRNAs and increasing the expression of autophagy signaling elements such as ULK1 and ATG7. This shift in signaling from apoptosis to autophagy ultimately overwhelms the 5-FU-induced apoptotic pathway, thereby mediating resistance to 5-FU treatment.170 Additionally, BIRC3, a member of the inhibitor of apoptosis protein family, inhibits apoptosis by directly inhibiting the caspase cascade.171 F. nucleatum can upregulate BIRC3 in CRC cells via the TLR-4/NF-kB pathway, leading to the suppression of tumor cell apoptosis and resistance to 5-FU chemotherapy 169 (Figure 6).
Mechanisms by which the gut microbiota regulates apoptosis pathways. Fusobacterium nucleatum promotes the expression of the autophagy signaling elements ATG7 and ULK1 and inhibits apoptosis through the activation of TLR-4. This bacterium also upregulates the anti-apoptotic protein BIRC3 via TLR-4, further suppressing apoptosis. The microbiota metabolite butyrate induces mitochondrial oxidative stress, promoting mitophagy, which can increase apoptosis. Additionally, butyrate functions as an HDAC inhibitor, further facilitating apoptosis. HDACi, histone deacetylase inhibitor; ROS, reactive oxygen species; TLR, Toll-like receptor.
F. nucleatum promotes anti-apoptotic effects in colon cancer cells by releasing extracellular heptose-related metabolites.172 ADP-heptose (ADP-H) and heptose-1, 7-bisphosphate are produced by gram-negative bacteria that trigger the ALPK1/TIFA pathway, leading to NF-κB activation.173 F. nucleatum releases an ADP-H-related molecule into its microenvironment, which exhibits the biological characteristics of ADP-H, an ALPK1 ligand, and potently activates NF-κB in intestinal epithelial cells via the ALPK1/TIFA/TRAF6 pathway. Furthermore, F. nucleatum releases butyrate, which synergizes with ADP-H in the ALPK1-dependent activation of NF-κB, thereby increasing the expression of the inflammatory cytokine IL-8 and two anti-apoptotic genes, BIRC3 and TNFAIP3, ultimately promoting CRC cell survival and reducing the chemosensitivity to 5-FU in vitro.
In contrast, the gut microbial metabolite butyrate potentiates 5-FU efficacy against CRC through distinct pro-apoptotic mechanisms. Butyrate preferentially accumulates within CRC cells owing to their reliance on glycolysis (Warburg effect),174 unlike normal colonocytes, which utilize butyrate as an energy source.175 As an endogenous histone deacetylase inhibitor, intracellular butyrate modulates the Akt/ERK and SMAD3 signaling pathways,126^,^176 enhancing apoptosis and 5-FU sensitivity.177 Furthermore, sodium butyrate (NaB) triggers ROS-mediated mitophagy linked to apoptosis via the PINK1/Parkin pathway178 (Figure 6). Consistent with these effects, the combination of 5-FU and NaB yielded synergistic antiproliferative effects in vitro and in vivo. This combination therapy can also foster a healthier gut microbial composition by enriching beneficial taxa (e.g., Bacteroidetes, Ligilactobacillus, and butyrate/acetate producers), potentially alleviating 5-FU toxicity. The chemosensitizing properties of butyrate may be extended to drugs such as gemcitabine for pancreatic cancer, although the specific mechanisms require further investigation.53
A recent study further explored the mechanisms underlying NaB-induced apoptosis, demonstrating that NaB enhances the 5-FU-mediated growth inhibition and apoptosis of gastric cancer cells by directly downregulating TS expression.179 Specifically, NaB downregulated the expression of key genes involved in DNA synthesis (TYMS and TK1), DNA replication (MCM4 and RRM2), cell cycle progression (CCNE1 and CCNA2), cell proliferation (FOXM1 and c-Myc), and stress responses (SIRT1 and DNAJA1), leading to cell cycle arrest and suppression of DNA replication. Notably, TYMS encodes TS, the target enzyme of 5-FU, whereas TK1 is critical for the recovery of dTMP synthesis. Downregulation of both enzymes attenuates tumor cell resistance to 5-FU. Furthermore, NaB induces apoptosis by disrupting mitochondrial energy metabolism and ATP synthesis. Moreover, sodium propionate exerts similar effects and mechanisms as those of NaB, including the modulation of TS, FOXM1, and MCM4.180 These findings suggest that the sodium salts of gut microbiota-derived SCFAs possess pro-apoptotic effects and potentiate the efficacy of antimetabolite drugs. However, this hypothesis requires further validation through additional research.
Microbial biomarkers for predicting the response to antimetabolite drugs
Emerging evidence indicates that gut microbiota profiles can predict patient responses to antimetabolite drugs such as MTX in RA (Table 2). A key study demonstrated that the baseline gut microbial taxonomic composition and functional potential in treatment-naive patients with RA were significantly correlated with the subsequent clinical response to MTX.181 Good responders (GR) exhibited lower microbial diversity than poor responders (PR). The PR group was characterized by a higher F/B ratio and enrichment of specific taxa (e.g., Euryarchaeota, Escherichia/Shigella), while the GR group showed higher relative abundances of Bacteroides and Prevotella. Functionally, patients with PR had enriched pathways related to MAPK signaling, DNA replication, and fatty acid degradation. In contrast, patients with GR had diminished pathways involving LPS metabolism and folate biosynthesis. By leveraging these metagenomic features, a random forest machine learning model successfully predicted MTX response status with 80% accuracy in an independent validation cohort (n = 21).181
A distinct predictive approach categorized patients with RA into enterotypes based on the dominant bacterial genera.183 Functional gene analysis revealed the enrichment of glycan synthesis and metabolism pathways in the Prevotella-dominant enterotype (RA E1), in contrast to the enrichment of vitamin and peptidoglycan synthesis pathways in the Bacteroides-dominant enterotype (RA E2). Clinically, patients belonging to the RA E1 enterotype demonstrated significantly better responses to MTX treatment than those in the RA E2 group.183
Gut microbial metabolic activity is associated with MTX efficacy in psoriasis.182 Post-MTX treatment, the GR group showed reduced organic and fatty acid levels, whereas the PR group showed elevated carbohydrates and amino acids, indicating that differential metabolism is associated with treatment outcomes. Mirroring the findings in RA,181 higher microbial diversity was observed in the PR group, potentially signifying greater resistance to MTX-induced changes. Specific taxa, including Bacilli, Lactobacillales, and unclassified Burkholderiales members, were enriched in the PR group. Distinct microbial functional signatures were also apparent; GR microbiomes showed increased activity in arginine/polyamine biosynthesis, purine degradation, and phosphatidylglycerol synthesis pathways, but decreased mannan degradation. Conversely, notable pathway alterations in PR microbiomes were mainly limited to methylerythritol phosphate and methylglyoxal degradation.182
Microbial metabolites are emerging predictors of sensitivity to chemotherapy. For example, differences in the gut microbiota between high-fat diet-induced obese and lean mice influence tumor responses to gemcitabine/paclitaxel, with lean mice showing better responses. This effect was microbiota dependent, as demonstrated by FMT experiments,185 which revealed distinct microbial metabolic outputs with obese mice showing enrichment in queuosine biosynthesis and lean mice favoring SAM metabolism. Queuosine promotes chemoresistance, which may involve upregulation of peroxiredoxin 1. In contrast, the antitumor metabolite SAM enhances chemosensitivity, with SAM administration improving treatment responses in obese mice.185 Similarly, in diabetic mouse models, the enrichment of menaquinol-producing bacteria was correlated with chemoresistance,186 likely due to its antioxidant capacity, which protects against drug-induced ROS. These findings highlight the regulation of chemotherapy outcomes by microbiota-derived metabolites such as queuosine, SAM, and menaquinol.
Strategies to improve antimetabolite drug response via the gut microbiota
An increasing number of chemical adjuvants and interventions, namely prebiotics, probiotics, synbiotics, postbiotics, dietary intervention, and FMT, have been developed to enhance the efficacy of antimetabolites and mitigate their toxicity (Table S1).
Supplementation with prebiotics, probiotics, synbiotics, or postbiotics
5.1.
Prebiotics
5.1.1.
Prebiotics, as defined by the International Scientific Association of Probiotics and Prebiotics, are substrates selectively utilized by host microorganisms to confer health benefits.187 Numerous prebiotics have been identified that may offer benefits during antimetabolite drug therapy. These are primarily derived from plant and animal sources, encompassing carbohydrate molecules (e.g., astragalus polysaccharides and fructooligosaccharides (FOSs)) and noncarbohydrate compounds (e.g., cryptotanshinone and casuarinin). Synthetic prebiotic mixtures, such as certain fiber blends, represent a smaller category (Table S1).
Natural carbohydrate prebiotics (polysaccharides and oligosaccharides) derived from diverse sources modulate the gut microbiota to improve antimetabolite therapy outcomes. Several studies have demonstrated their benefits, particularly in the mitigation of 5-FU toxicity. Oyster polysaccharide (CHP) and alginate oligosaccharide (AOS) both alleviate 5-FU–induced enteritis by enriching beneficial bacteria such as Akkermansia, increasing SCFA production, enhancing gut barrier integrity (AOS via MLCK signaling), and reducing inflammation (CHP via TLR signaling, AOS via TLR4/MyD88/NF-κB inhibition and apoptosis modulation).24^,^26 Polysaccharides from Astragalus, Poria, and Albuca bracteata showed similar potential (Table S1). FOS, alone or with arginine, specifically target 5-FU–induced mucositis by boosting SCFAs, preserving barrier function, and promoting beneficial genera such as Bacteroides and Lactobacillus 188^,^189.
In addition to mitigating toxicity, prebiotics such as xylan can also be used for drug delivery.190 Common drug delivery strategies include liposomes, polymeric nanoparticles, lipid nanoparticles, inorganic nanoparticles, dual targeting nanoparticles, and exosomes.191 Xylan–stearic acid nanoparticles (SCXN) carrying capecitabine improved drug delivery to CRC tumors and enhanced survival in mice compared to free drugs.192 This system simultaneously modulated antitumor immunity (favoring dendritic cell maturation and CD8+ T cell responses over Tregs) and reshaped the gut microbiome toward a beneficial state (enriching Clostridia, Lachnospiraceae, and Akkermansia; suppressing Desulfovibrio; and boosting SCFAs).192 These effects highlight its multifaceted therapeutic potential.
A recent study developed 5-FU–loaded liposomes by combining the prebiotic xylan derivative Sxy with 1, 2-dipalmitoylphosphatidy-lethanolamine, a homolog of A. muciniphila active phospholipid. This delivery system enhanced CRC treatment efficacy via a mechanism similar to that of SCXN.193
In addition to carbohydrates, non-carbohydrate natural extracts can modulate the gut microbiota to enhance antimetabolite therapy. For example, cryptotanshinone (CTS), derived from Salvia miltiorrhiza, ameliorates 5-FU–induced colitis in mouse models by regulating microbial lipid metabolism that involves shifting specific bacterial populations (e.g., Muribaculaceae and Lactobacillus) and normalizing host lipid markers.194 CTS can also activate the PI3K/Akt pathway to exert positive effects.195 Another Salvia compound, dihydrotanshinone (DHTS), combats 5-FU mucositis by restoring microbiota balance, enriching Akkermansia, and suppressing systemic inflammatory markers such as IL-6 and TNF-α 196. Patchouli alcohol, from Pogostemon cablin, supports intestinal barrier integrity, partly by influencing microbiota structure and inhibiting TLR2/MyD88/NF-κB signaling, thereby preventing enteritis.18 Curcumin has also demonstrated promising prospects for treating ulcerative colitis.197 Other non-carbohydrate compounds such as berberine,12^,^34^,^198 casuarinin,199 and saikosaponin-A38 also leverage microbiota interactions to improve antimetabolite outcomes (Table S1).
Probiotics
5.1.2.
Probiotics are live microorganisms that, when administered in adequate quantities, confer health benefits to the host 200 and can markedly influence outcomes during chemotherapy, including 5-FU and MTX treatments. Specific strains, such as L. casei var. rhamnosus and L. reuteri DSM 17938 mitigated 5-FU mucositis by modulating the microbiota balance.201 Pediococcus pentosaceus PP34 combats 5-FU toxicity by reducing oxidative stress and inflammation, concurrently altering gut microbial composition (e.g., enriching Akkermansia).202 A. muciniphila specifically protects against MTX intestinal injury and is linked to the maintenance of gut homeostasis and stem cells via Wnt signaling and SCFA production.203 The yeast Saccharomyces boulardii (CNCM I-745) exerts broad effects, improving bone/joint health, reducing inflammation in MTX-treated arthritis models,204 and alleviating 5-FU mucositis by limiting inflammation and restoring gut permeability.205
Probiotic mixtures also show promise: Lactobacillus/Bifidobacterium combinations regulated inflammation in 5-FU–treated CRC models (via NF-κB inhibition and mucin preservation),206 and L. casei Shirota/B. breve combinations enhanced antitumor immunity during gemcitabine/cisplatin treatment via the gut-tumor axis in bladder cancer models.54 A controlled clinical trial demonstrated that for psoriasis patients, supplementing MTX therapy by adding probiotics (Bifidobacterium longum, L. acidophilus, and Enterococcus Capsules) helped maintain intestinal barrier integrity more effectively than MTX alone.207 Serum levels of calprotectin and zonulin did not substantially increase in the combination group, unlike those in the MTX-only control group. A novel “intestinal–vaginal” synergistic drug delivery strategy combines oral B. longum subsp. longum NCU-05 with intravaginal Lacticaseibacillus crispatus NCU-28.208 This strategy enhanced the anticancer efficacy of 5-FU by activating the p53 signaling pathway and upregulating pro-apoptotic protein expression. It simultaneously restored intestinal and vaginal microbial homeostasis, thereby mitigating inflammation-related tissue damage at both sites.
Synbiotics
5.1.3.
Defined as a mixture of live microorganisms and substrate(s) selectively utilized by host microorganisms that confer health benefits to the host,209 synbiotics offer strategies to counteract chemotherapy complications. One example involves encapsulating specific Bifidobacterium, Lactobacillus, Streptococcus, and Clostridium strains in resistant starch; this synbiotic markedly reduced 5-FU–induced inflammation (cytokines, NF-κB signaling) while preserving microbial abundance and composition, thus mitigating dysbiosis.27 Another study used a combination of several Lactobacillus and Bifidobacterium strains with prebiotics (inulin and lactoferrin) to treat gemcitabine/nab-paclitaxel–induced dysbiosis in a pancreatic cancer model. This synbiotic increases microbial richness and SCFA levels, consequently reducing chemotherapy side effects and cancer-associated stromatogenesis.210 A multicenter randomized clinical trial included 61 patients with esophageal cancer receiving neoadjuvant chemotherapy (including 5-FU).211 This trial found that patients experienced a considerable reduction in adverse events when supplemented with synbiotics (containing L. casei Shirota and galacto-oligosaccharides) compared to the control group. This beneficial effect is attributed to the ability of synbiotics to enhance the abundance of beneficial gut bacteria and elevate the concentrations of SCFAs, specifically acetic and propionic acids, thereby mitigating the incidence of chemotherapy-induced febrile neutropenia.
Postbiotic
5.1.4.
Postbiotics, defined as preparations of inanimate microorganisms and/or their components that confer health benefits to the host,212 can potentially augment chemotherapy outcomes. For example, L. rhamnosus–derived postbiotics show promise: the cell-free supernatant from the strain GG displays selective anticancer activity and enhances 5-FU sensitization,213 whereas an inactivated preparation of the strain CGMCC1.3724 mitigates 5-FU–induced mucosal damage via mechanisms involving structural preservation, reduced inflammation, enhanced mucus production, and increased Treg populations.214 Microbial metabolites also function as postbiotics. For instance, UroB possesses potent immunomodulatory properties including boosting natural killer cell activity, inhibiting Tregs, downregulating PD-L1 expression, upregulating antigen presentation machinery (HLA-B and TCR), and enhancing overall antigen presentation. In combination with capecitabine, UroB regulates the gut microbiota and suppresses CRC tumor growth in mice.215
Dietary intervention
5.2.
Dietary intervention to modify the gut microbiota can increase the efficacy of antimetabolite drugs. These interventions include dietary restriction (DR), fiber-rich diet, and ketogenic diet.
The benefits of DR on the efficacy of antimetabolite drugs remain controversial. Previous studies demonstrated that DR may benefit the prognosis of patients undergoing chemotherapy.216 Research has shown that 30% DR for two weeks before chemotherapy markedly improves overall survival in both young and old mice treated with 5-FU.217 DR prevents the loss of lysozymes and increases the abundance of Lactobacillus, thereby inhibiting the translocation of opportunistic pathogens into the gut. This effect was more prominent in older mice.217 Given that elderly patients often exhibit an increased abundance of opportunistic pathogens in their feces, DR holds considerable potential for improving the safety of and tolerance to chemotherapy in older individuals.217 Moreover, 30% DR for two weeks markedly improved the survival rates after exposure to a lethal dose of MTX. DR alleviates intestinal inflammation by increasing the abundance of Lactobacillus and preserving the number of basal crypt proliferating cell nuclear antigen-positive cells, thus protecting intestinal stem cell function.45 However, another study did not replicate these findings. It revealed that although 48 h of DR before treatment slowed epithelial proliferation and increased microbial diversity and richness, it did not affect the abundance of Lactobacillus. Furthermore, the beneficial effects of DR are overshadowed by its negative impact on body weight, resulting in a failure to effectively alleviate MTX-induced gastrointestinal mucositis.218 Therefore, exploring more rational DR strategies may be crucial for optimizing the effects of DR.
A fiber-rich diet in mice undergoing 5-FU treatment promoted the production of microbial metabolites and reduced neuroinflammation.219 This dietary intervention substantially altered the gut microbiota composition. It increased the abundance of Bacteroidaceae and Akkermansiaceae that correlated with increased propionate production, reduced astrocyte density, and subsequent alleviation of neuroinflammation.219 Furthermore, a whey-based diet containing medium-chain triglycerides (MCTs) has demonstrated beneficial effects against MTX-induced gastrointestinal damage.220 The readily absorbed MCTs provided rapid energy and facilitated the renewal and repair of intestinal epithelial cells. This dietary approach also modified the gut microbiota structure and activity, leading to an increase in branched-chain fatty acid levels and a reduction in MTX-induced gastrointestinal mucositis.220
FMT
5.3.
FMT is used to directly alter a recipient’s gut microbiota to normalize its composition and derive therapeutic benefits.221 Clinically, FMT has been used to treat recurrent C. difficile infections, irritable bowel syndrome, and IBD, among other conditions.222 Importantly, the safety of FMT remains a challenge, with common adverse events including vomiting, abdominal pain, diarrhea, and infection.223 Screening of healthy donors is key to the success of FMT.224 Recent findings suggest that FMT can directly manipulate the gut microbiota and improve chemotherapy-induced mucositis. For example, FMT improved intestinal mucosal damage in a mouse model of colon cancer treated with FOLFOX chemotherapy (5-FU, oxaliplatin, etc.).31 Specifically, FMT reduced the severity of diarrhea and intestinal mucositis, inhibited the TLR/MyD88/NF-κB signaling pathway and apoptosis, and restored disrupted fecal microbiota composition while avoiding bacteremia. This resulted in the safe and effective alleviation of FOLFOX-induced intestinal mucositis in tumor-bearing mice.31 In another study, FMT markedly reversed the disruption of gut microbiota caused by ampicillin and/or 5-FU in mice.225
In contrast to the results observed for 5-FU, FMT did not exert the expected effects during MTX treatment. An exploratory randomized placebo-controlled trial revealed that the treatment failure rate of FMT in patients with active peripheral psoriatic arthritis receiving MTX was substantially higher than that in the placebo control group.226
Others
5.4.
Exogenous folate supplementation enhances the therapeutic efficacy and reduces the toxicity of MTX. Notably, the gut microbiota of patients with RA exhibit overexpression of metabolic pathways involved in folate biosynthesis. The MTX-induced inhibition of folates has been proposed to reverse this microbial overproduction, potentially increasing microbiome α-diversity, which could benefit the gut microbiota of patients with RA.40^,^81 However, these beneficial effects on the gut microbiota tend to diminish with prolonged folate deprivation, necessitating folate supplementation in cases of MTX overdose.81 Given that the toxicity of MTX is linked to its antifolate properties,227 folate or its derivatives may be used to prevent the toxic side effects and MTX retention.46^,^72 In addition to counteracting MTX-mediated inhibition of DHFR, folate mitigates MTX side effects by modulating the gut microbiota.46 Specifically, the folate derivative leucovorin increases the abundance of Bifidobacterium, which strengthens the intestinal mucosal barrier and exerts anti-inflammatory effects, thereby alleviating MTX-induced gastrointestinal toxicity.46
Magnesium isoglycyrrhizinate treatment markedly alleviates MTX-induced intestinal and liver damage; as such, it is routinely used as a hepatoprotective agent in clinical settings to treat DILI.228 It alters the composition of the gut microbiota by increasing the abundance of the beneficial bacterium, Lactobacillus and decreasing Muribaculaceae. This modulation helps to remodel the intestinal barrier and inhibits bacterial translocation to the liver, ultimately reducing MTX-related DILI.50
Future directions
Current research on the complex interactions between antimetabolite drugs and the microbiome has some limitations. A primary challenge is the predominant reliance on cell and animal models with a scarcity of high-quality clinical cohort studies, which limits the strength of evidence. Furthermore, most studies utilize fecal microbiota as a proxy for the entire gut microbiome, despite the limited understanding of compositional variations across different gut regions. The prevalent use of 16S rRNA gene sequencing restricts the depth of the information obtained. Additionally, many studies lack dynamic microbiome profiling during the experimental processes, often comparing only pre- and post-intervention states. The variability in animal models, drug dosages, and dosing intervals further complicates cross-study comparisons. Finally, research on non-bacterial components of the microbiota, such as fungi and viruses, remains insufficient. Future investigations, considering the heterogeneity of patient populations and the increasing use of multidrug regimens, should employ advanced sequencing techniques, such as metagenomics, and incorporate dynamic monitoring of detailed alterations in both bacterial and non-bacterial microbes to yield more comprehensive and precise insights.
Conclusions
The bidirectional interactions between antimetabolite drugs and the gut microbiota are central to optimizing clinical outcomes. Future research should prioritize leveraging advanced tools including metagenomics and real-time microbial community monitoring to fully elucidate the underlying mechanisms of these interactions. These insights will enable the development of reliable microbial biomarkers for treatment response prediction and innovative microbiota-manipulation strategies. Additionally, future studies must account for patient diversity and combination therapies to translate these insights into precise, clinically applicable antimetabolite regimens.
Supplementary Material
Certificate_of_editing_FRTJD_1_2_4wqhms6lbt.pdfCertificate_of_editing_FRTJD_1_2_4wqhms6lbt.pdf
Supplementary_tables clean.docxSupplementary_tables clean.docx
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. 2020;206:107447. doi: 10.1016/j.pharmthera.2019.107447.31756363 · doi ↗ · pubmed ↗
- 2Prasad P, Verma S, Surbhi, Ganguly NK, Chaturvedi V, Mittal SA. Rheumatoid arthritis: advances in treatment strategies. Mol Cell Biochem. 2023;478(1):69–88. doi: 10.1007/s 11010-022-04492-3.35725992 · doi ↗ · pubmed ↗
- 3Zhou B, Xia X, Wang P, Chen S, Yu C, Huang R, Zhang R, Lu L, Yuan F, Tian Y, et al. Induction and amelioration of methotrexate-induced gastrointestinal toxicity are related to immune response and gut microbiota. E Bio Medicine. 2018;33:122–133. doi: 10.1016/j.ebiom.2018.06.029.30049384 PMC 6085585 · doi ↗ · pubmed ↗
- 4Chen YC, Hou CY, Hsu MH, Huang LT, Hsiao CC, Sheen JM. The impact of gut microbiota changes on methotrexate-induced neurotoxicity in developing young rats. Biomedicines. 2024;12(4):908. doi: 10.3390/biomedicines 12040908.38672262 PMC 11048417 · doi ↗ · pubmed ↗
- 5Li S, Zhu S, Yu J. The role of gut microbiota and metabolites in cancer chemotherapy. J Adv Res. 2023;64:223–235. doi: 10.1016/j.jare.2023.11.027.38013112 PMC 11464465 · doi ↗ · pubmed ↗
- 6Fernandes MR, Aggarwal P, Costa RGF, Cole AM, Trinchieri G. Targeting the gut microbiota for cancer therapy. Nat Rev Cancer. 2022;22(12):703–722. doi: 10.1038/s 41568-022-00513-x.36253536 · doi ↗ · pubmed ↗
- 7Ting NL, Lau HC, Yu J. Cancer pharmacomicrobiomics: targeting microbiota to optimise cancer therapy outcomes. Gut. 2022;71(7):1412–1425. doi: 10.1136/gutjnl-2021-326264.35277453 PMC 9185832 · doi ↗ · pubmed ↗
- 8Panebianco C, Andriulli A, Pazienza V. Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies. Microbiome. 2018;6(1):92. doi: 10.1186/s 40168-018-0483-7.29789015 PMC 5964925 · doi ↗ · pubmed ↗