Exploring T. cruzi IMPDH as a promising target through Chagas Box screening and AVN-944 inhibition
Angel Lobo-Rojas, Letícia Marchese, Amanda G. Eufrasio, Gabriel T. D. Souza, Michelle F. Catelli, Jessica D. N. Faria, Artur T. Cordeiro

TL;DR
Researchers explore T. cruzi IMPDH as a new drug target for Chagas disease and find AVN-944 to be a promising treatment candidate.
Contribution
The study identifies TcIMPDH as a druggable target and demonstrates AVN-944's efficacy against Chagas disease in cell-based assays.
Findings
AVN-944 inhibits TcIMPDH with submicromolar IC50 and outperforms benznidazole in infected cardiomyoblasts.
TcIMPDH shows conserved catalytic and allosteric residues, supporting its druggability.
TCMDC-143376 is identified as structurally similar to clinical IMPDH inhibitors.
Abstract
Chagas disease, caused by Trypanosoma cruzi, remains a leading cause of heart failure in Latin America, with current treatments limited to acute-phase efficacy, significant toxicity, and emerging resistance. Inosine monophosphate dehydrogenase (IMPDH) is an essential enzyme in guanine nucleotide salvage pathway and represents a promising alternative target. Here, we combined computational screening, biochemical and cell-based phenotypic assays that support T. cruzi IMPDH (TcIMPDH) as a druggable target and identify repurposing opportunities among clinical-stage inhibitors. Using Tanimoto similarity scoring against the library of 222 Chagas Box compounds, we identified TCMDC-143376 as uniquely similar to the clinical IMPDH inhibitors merimepodib and AVN-944. Phylogenetic analysis and multiple sequence alignment confirmed conservation of both catalytic and allosteric residues—drawn from…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
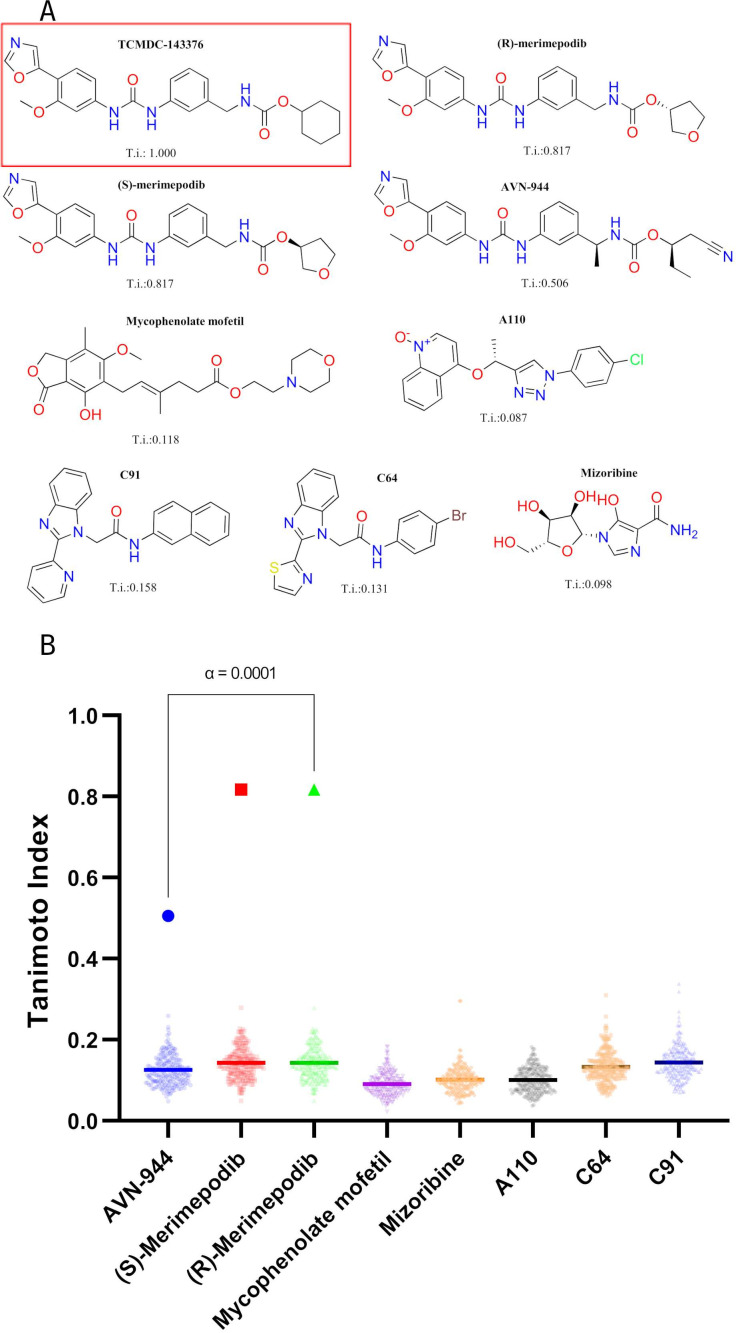 Fig 1
Fig 1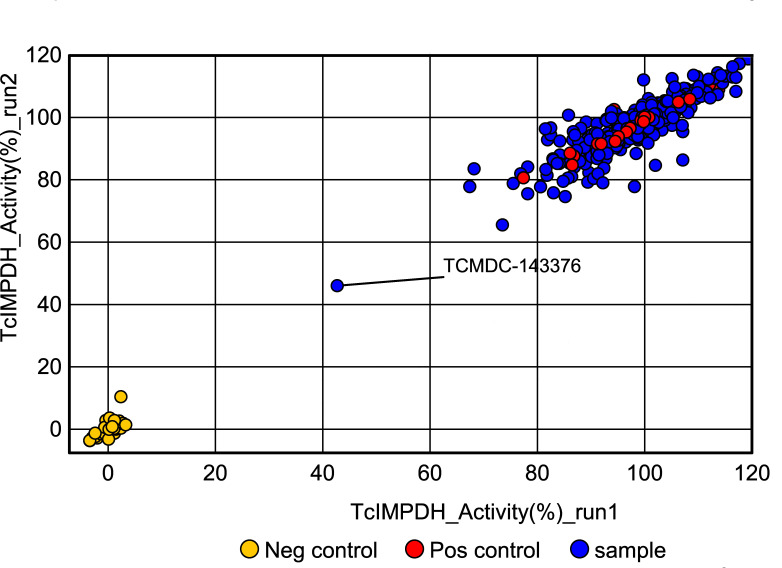 Fig 2
Fig 2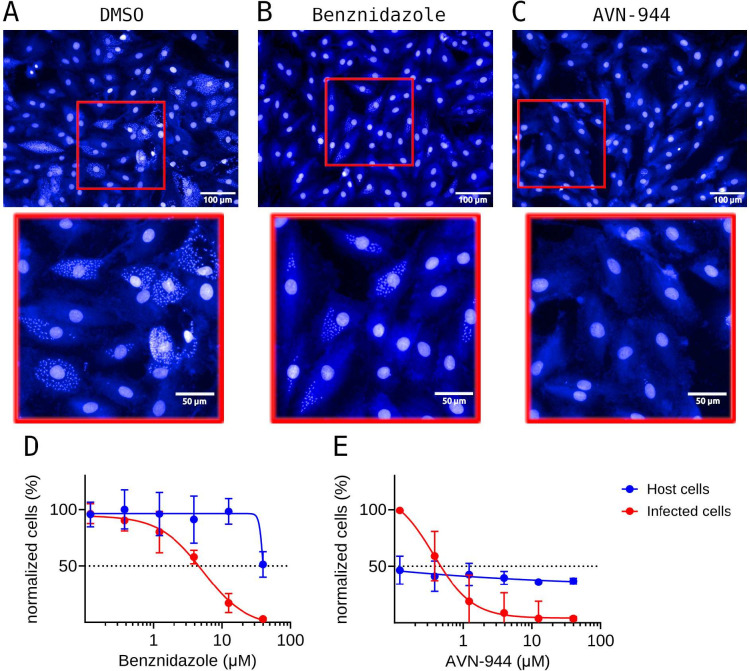 Fig 3
Fig 3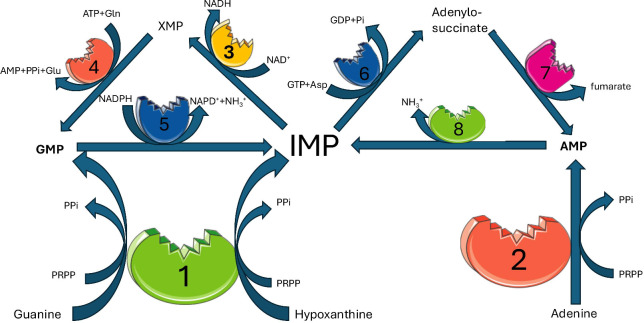 Fig 4
Fig 4| Compound name | IC50 (μM) | Confidence interval (95%) | R2 |
|---|---|---|---|
| TCMDC-143376 | 4.00 | 3.62…4.52 | 0.973 |
| Mycophenolate mofetil | 4.40 | 2.78…6.60 | 0.969 |
| AVN-944 | 0.20 | 0.18...0.25 | 0.984 |
| (S)-Merimepodib | 0.21 | 0.19...0.24 | 0.993 |
| (R)-Merimepodib | 0.37 | 0.32...0.42 | 0.994 |
| Compound name | EC50 (μM) | Confidence interval (95%) | R2 |
|---|---|---|---|
| Benznidazole | 3.00 | 1.76...3.65 | 0.928 |
| TCMDC-143376 | 15.9 | ||
| Mycophenolate mofetil | 1.50 | 0.40...2.98 | 0.933 |
| (S)-Merimepodib | 4.80 | 1.25...10.3 | 0.947 |
| (R)-Merimepodib | 3.00 | 1.17...5.49 | 0.913 |
| AVN-944 | 0.40 | 0.08…1.14 | 0.962 |
- —Financiadora de Estudos e Projetoshttp://dx.doi.org/10.13039/501100004809
- —Fundação de Amparo à Pesquisa do Estado de São Paulohttp://dx.doi.org/10.13039/501100001807
- —Fundação de Amparo à Pesquisa do Estado de São Paulohttp://dx.doi.org/10.13039/501100001807
- —Fundação de Amparo à Pesquisa do Estado de São Paulohttp://dx.doi.org/10.13039/501100001807
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBiochemical and Molecular Research · Trypanosoma species research and implications · Enzyme Structure and Function
INTRODUCTION
Chagas disease, caused by the protozoan parasite Trypanosoma cruzi, remains a major neglected tropical disease more than a century after its discovery (1). Endemic in Latin America and increasingly detected in non-endemic regions due to migration (2), Chagas disease affects an estimated 6-7 million people worldwide (3). Despite its public health burden, therapeutic options remain limited to just two frontline drugs: benznidazole and nifurtimox, both introduced over 50 years ago (4).
Benznidazole, the first-line treatment, induces DNA damage in T. cruzi; nonetheless, the persistence of rare, non-replicative amastigotes in infected tissues has been observed (5). Indeed, resistance to benznidazole can develop readily within a single population, by independently acquiring mutations in the gene encoding a mitochondrial nitroreductase, which can give rise to distinct drug-resistant clones within a single population (6). Nifurtimox, a nitrofuran prodrug, has been used for over 40 years to treat Chagas disease. Its mechanism of action was initially linked to oxidative stress, but newer evidence points to activation by a parasite-specific type I nitroreductase (7). The resulting metabolite, a reactive open-chain nitrile, lacks selectivity, affecting both parasite and host cells, producing important side effects. Despite their different mechanisms, both nifurtimox and benznidazole are mainly effective in the acute phase. However, due to limited diagnostics and healthcare infrastructure in endemic regions, infections often go unnoticed until the chronic phase, where current drugs show poor efficacy. This underscores the urgent need for new therapeutic targets and treatments.
Alternative drug targets have been explored, including T. cruzi CYP51, a sterol 14α-demethylase essential for parasite viability (8, 9). Despite initial promise, clinical trials targeting CYP51 were halted due to suboptimal efficacy (10) and potential host toxicity (11). Cytochrome b, a component of complex III in the mitochondrial electron transport chain, also has emerged as a druggable target in Trypanosoma cruzi and Leishmania spp. by phenotypic screening assays, with inhibitors like GNF7686 acting at the Qi site, blocking respiration and parasite growth (12). However, its mitochondrial maxicircle-encoded nature makes it highly prone to resistance through point mutations, such as the L198F mutation in GNF7686-resistant T. cruzi (13). Due to this high resistance potential, cytochrome b is now deprioritized in some drug discovery pipelines using resistant strain counterscreens (14, 15). On the other hand, proteasome inhibitors have emerged as promising anti-kinetoplastid agents, with compounds like GNF6702 demonstrating unprecedented in vivo efficacy, successfully clearing parasites in multiple mouse models (16). This azabenzoxazole derivative acts as a non-competitive inhibitor of the kinetoplastid proteasome and laid the groundwork for potent analogs, such as LXE408, a next-generation inhibitor characterized by high-resolution cryo-EM in complex with Leishmania tarentolae proteasomes, has progressed to clinical trials (17). Despite these advances, challenges related to toxicity and selectivity remain, and no proteasome inhibitor has yet reached widespread clinical use for Chagas disease. These limitations highlight the continued need to explore alternative drug targets with safer and more effective therapeutic profiles.
One promising avenue lies in the purine salvage pathway, which is essential for T. cruzi survival due to its inability to synthesize purines de novo (18, 19). Instead, Trypanosomatids rely on salvaging purines from external sources. For instance, it has been previously observed that epimastigotes uptake hypoxanthine from LIT (Liver Infusion Tryptose) medium during its exponential growth phase (20). In this pathway participate enzymes, such as adenine phosphoribosyltransferase (APRT), hypoxanthine-guanine phosphoribosyltransferase (HGPRT), inosine-5′-monophosphate dehydrogenase (IMPDH), and guanosine monophosphate synthase (GMPS). The first three enzymes have been identified in proteomic studies in T. cruzi glycosomes (21), and the last one has been demonstrated to be essential in the related organism T. brucei (22). In particular, IMPDH plays a critical role by catalyzing the NAD^+^-dependent oxidation of inosine monophosphate (IMP) to xanthosine monophosphate (XMP), the key precursor for guanine nucleotide biosynthesis (23). While mammals possess IMPDH enzymes as well, parasite-specific differences in structure and regulation may offer opportunities for selective inhibition. It is worth noting that IMPDH has been explored as a drug target in pathogenic prokaryotic organisms; it has been validated as a promising drug target in pathogens like Helicobacter pylori (24, 25), Bacillus anthracis, Staphylococcus aureus, and Listeria monocytogenes (26). Structural determinants of inhibitor selectivity have been identified in bacterial IMPDHs (24). Even in another protozoan parasite, the apicomplexan Cryptosporidium parvum, IMPDH has also been studied as a drug target (27). The C. parvum IMPDH is closely related to the prokaryotic counterparts (28), and efforts are underway to repurpose Cryptosporidium IMPDH inhibitors as broad-spectrum antimicrobials (26).
IMPDH inhibitors for clinical use have been developed and explored for therapeutic purposes, ranging from immunosuppressants to antivirals and anticancer agents. Mycophenolic acid (MPA), an FDA-approved uncompetitive IMPDH inhibitor, is widely used as an immunosuppressant to prevent organ transplant rejection (29). By selectively inhibiting IMPDH, it interferes with the de novo synthesis of guanosine nucleotides, while most cell types can compensate via purine salvage pathways, T and B lymphocytes are particularly dependent on de novo purine synthesis, making them susceptible to IMPDH inhibition. As a result, MPA exerts potent cytostatic effects on lymphocytes, suppresses antibody production, and interferes with glycoprotein glycosylation involved in immune cell adhesion (30, 31). Its FDA-approved prodrug, mycophenolate mofetil, offers improved oral bioavailability, avoiding some side effects but retaining the same mechanism of action (32). Noteworthy, clinical evidence shows that mycophenolate mofetil increases the risk of Chagas disease reactivation in heart transplant patients (33), also corroborated in mice revealed that although it reduces parasitemia, it does not improve survival during acute T. cruzi infection (34). Another FDA-approved drug, ribavirin, exerts antiviral effects partly through IMPDH inhibition but also directly targets viral RNA polymerases (35). More recent efforts have yielded second-generation IMPDH inhibitors, such as VX-148, a potent IMPDH inhibitor (36) whose derivatives, AVN-944 and merimepodib, have progressed to clinical trials phase II. AVN-944, originally developed for cancer due to its capacity to deplete guanine nucleotides, inhibits ribosomal RNA synthesis and leads to nucleolar disruption, inducing apoptosis via cellular stress responses (37, 38). Similarly, merimepodib, initially developed as an immunosuppressant (39), has also demonstrated broad-spectrum antiviral activity, including efficacy against Zika virus (40) and hepatitis C virus (41), further underscoring the therapeutic versatility of IMPDH inhibitors. Finally, mizoribine, a ribavirin derivative, is under investigation for rheumatoid arthritis (42). Collectively, these compounds highlight the clinical relevance of IMPDH inhibition across diverse therapeutic areas and reinforce the rationale for repurposing such agents for neglected diseases like Chagas disease.
In this study, we focus on the T. cruzi IMPDH as a druggable target. Using a ligand-based drug discovery approach, we identified potential inhibitors within the Chagas Box compound collection, a curated, open-source library of chemically diverse, drug-like molecules with confirmed activity against T. cruzi, made publicly available to accelerate Chagas disease drug discovery (43). Notably, we identified TCMDC-143376, which showed structural similarity to AVN-944 and Merimepodib, suggesting potential cross-species inhibitory effects. Biochemical and phenotypic assays confirmed that both AVN-944 and merimepodib are potent inhibitors of TcIMPDH and display significant antiparasitic activity against T. cruzi. These findings not only reinforce TcIMPDH as a promising and previously unexplored drug target for Chagas disease but also position AVN-944 and merimepodib as valuable proof-of-concept compounds for validating TcIMPDH inhibition as a viable therapeutic strategy.
MATERIALS AND METHODS
In silico similarity analysis between IMPDH inhibitors and Chagas Box compounds
To assess the potential structural similarity between known IMPDH inhibitors and compounds with activity against Trypanosoma cruzi, we performed a Tanimoto similarity analysis (44). The objective was to identify Chagas Box hits that share a high degree of molecular similarity with inhibitors previously developed for bacterial and human IMPDHs. The compounds used as queries were prokaryotic IMPDH inhibitors as C64, C91, A110 (24), mycophenolate mofetil (45), and human IMPDH inhibitors as mizoribine, merimepodib, and AVN-944.
These molecules were used as queries to calculate Tanimoto index against the 222 compounds Chagas Box set (43), all of them with confirmed activity against T. cruzi intracellular amastigotes. Molecular structures were processed as SMILES strings and similarity calculations were performed using the RDKit cheminformatics library in Python (46). Molecules were converted to Morgan fingerprints (radius = 2), and pairwise Tanimoto similarity indices were computed between each reference inhibitor and all Chagas Box compounds. Iterative Grubbs’ test (α = 0.0001) was used to identify outliers inside each population (47) and plotted using GraphPad Prism 9.5.0.
Bioinformatics and phylogenetic analysis
The IMPDH gene from T. cruzi was identified in the kinetoplastid genomic resource database (https://tritrypdb.org/tritrypdb/app) under the code TcCLB.507211.40, the translated sequence was retrieved as fasta format and used to perform a search by BLASTP (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (48), restricted search sets were used by inclusion and/or exclusion of certain organisms or phylogenetic groups sequences to assure adequate sampling of IMPDH genes from landmark organisms. To perform the multiple sequence alignment (MSA) IMPDH protein sequences from several organisms were used: T. cruzi (XP_805772.1, which is the product from TcCLB.507211.40), T. brucei (AAB46420.1) whose 3D structure was solved by following an in cellulo crystallization approach (49), Leishmania donovani (XP_003860332.1), two isoforms sequences from Homo sapiens 1 (NP_001136045.1) and 2 (NP_000875.2), two isoforms sequences from Mus musculus 1 (NP_001289862.1) and 2 (NP_035960.2), Plasmodium falciparum (XP_001352079.1), Arabidopsis thaliana (NP_178065.1), Escherichia coli (P0ADG7.1) and Tritrichomonas foetus (XP_068358496.1) whose x-ray solved structure is available at https://www.rcsb.org/ under the code 1MEW. Retrieved sequences in fasta format were aligned using the online program Clustal Omega (50), from the EMBL’s European Bioinformatics Institute (https://www.ebi.ac.uk/jdispatcher/msa/clustalo), and subsequently analyzed with the online server ESPript 3 (https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi) (51).
Cloning, heterologous expression, and purification of the T. cruzi IMPDH product
The IMPDH gene from T. cruzi was identified in the kinetoplastid genomic resource database (https://tritrypdb.org/tritrypdb/app) under the code TcCLB.507211.40, retrieved ORF sequence was sent to Genscript services in order to synthesize and clone into pET28a vector. Flanked with XhoI and BamHI restriction sites. The sequence of the gene was optimized by Genscript, taking into account a wide variety of factors that regulate and influence gene expression levels, and by GenSmart codon optimization tool. Taking advantage of the restriction enzyme sites, the gene was subcloned into the pETSUMO vector as previously made with other recombinant enzymes (52, 53). The resulting plasmid pETSUMO-TcIMPDH was used to transform BL21 by conventional procedures and overpressed by autoinduction (54) at 20°C, 200 RPM for 72 h. Bacterial cells were resuspended in buffer A (50 mM Tris-HCl pH 8.0 + 0.3 M KCl + 5% glycerol), and clear lysate was obtained by sonication and centrifugation. Thus, it was charged onto a Ni-sepharose affinity column previously equilibrated with buffer A. After washing steps, the recombinant protein was eluted with increasing concentrations of buffer B (50 mM Tris-HCl pH 8.0 + 0.3 M KCl + 5% glycerol + 1 M imidazole). Fractions containing the recombinant protein were pooled. His-SUMO tag was cleaved by incubation with ULP-1 protease at 4°C overnight. Afterward, a reverse affinity chromatography was made, and finally, fractions with TcIMPDH were pooled, added 10% glycerol and 1 mM DTT, and stored at −80 °C. Along purification steps, the protein concentration was assessed by Bradford method and the performance checked by SDS-PAGE (55).
Enzyme kinetic characterization
The Michaelis-Menten kinetic parameters (Km) for IMP and NAD^+^ were determined using the direct assay by following the formation of NADH fluorescence intensity (Ex = 340 nm; Em = 485 nm) at 25°C, in black 384-wells microplates. Fluorescence was recorded every 30 s over 10 min using a CLARIOstar plate reader (BMG LabTech) and steady-state velocities were calculated from the maximum slope of the fluorescence curve. All reactions were performed in triplicate, under optimized buffer conditions: 50 mM tri-sodium citrate (pH 7), 100 mM KCl, and 1 mM DTT. For both substrates, 1 μM of purified recombinant TcIMPDH was used. To determine the Km for IMP, NAD^+^ was held at a saturating concentration (1 mM), while IMP was tested in eight 1:2 serial dilutions starting from 1 mM. Conversely, to determine the Km for NAD^+^, IMP was fixed at 1 mM, and NAD^+^ was serially diluted in the same manner. Reactions were initiated by the addition of IMP, and data were analyzed using nonlinear regression to fit the Michaelis-Menten equation in GraphPad Prism 9.5.0.
Screening of Chagas Box library for TcIMPDH inhibitors
Compounds were assayed at a single concentration of 10 μM. They were provided by GSK (GlaxoSmithKline) in a 384-well ready-to-use assay microplate containing 25 nL of each sample at 10 mM (100% DMSO). Because some tested compounds showed fluorescence at wavelengths E_x_ 340 nm/E_m_ 460 nm which are used to detect direct product NADH of the reaction, the coupled assay with diaphorase and resorufin fluorescence (E_x_ 545 nm/E_m_ 600 nm) (52) was used and recorded every 30 s for 10 min. Velocities at steady state were calculated in the plate reader (CLARIOstar, BMG LabTech) as the maximum slope. Next, 20 μL of Mix solution containing 1.25 μM TcIMPDH, 0.250 mM NAD^+^, 10 μM resazurin in 62.5 mM citrate buffer pH 7.0, 125 mM KCl, and 1 mM DTT, 1.25 U/mL diaphorase was dispensed into wells and the reaction initiated by addition of 5 μL of 0.25 mM IMP. After 15 min, resorufin fluorescence (Ex = 570 nm; Em = 590 nm) was measured in a plate reader (CLARIOstar, BMG LabTech). Data were normalized and plotted using GraphPad Prism 9.5.0.
Measuring of half-maximal inhibitory concentration of compounds against TcIMPDH activity
To determine the concentration necessary to inhibit 50% of TcIMPDH activity (IC_50_), stock solutions (10 mM) of compounds mycophenolate mofetil, (S)-merimepodib, AVN-944, and (R)-merimepodib were serially diluted in DMSO, applying a 1⁄2-log dilution factor. The above-mentioned diaphorase-coupled assay was used. The final concentrations of components in the enzymatic assay were: 0.2 mM NAD^+^, 0.2 mM Inosine 5′-monophosphate (IMP), 1 μM TcIMPDH, 1 U/mL diaphorase, 10 μM resazurin in 50 mM citrate buffer pH 7.0 plus 100 mM KCl. The reaction was started by adding IMP. TcIMPDH activity was normalized, and the IC_50_ was calculated using GraphPad Prism 9.5.0.
Determining the half-maximal effective concentration (EC₅₀) against Trypanosoma cruzi amastigotes in H9c2 cells
To determine EC_50_ to kill intracellular amastigotes in H9c2 host cells, we used an image-based assay previously described in Alonso-Padilla et al. (56). Briefly, T. cruzi trypomastigotes (from LLC-MK2 supernatant) were used to infect H9c2 rat cardiomyocytes overnight. After washing, cells were incubated for 48 h in fresh DMEM, then seeded (1.5 × 10^3^ cells/well) into 384-well plates with 50 µL assay volume (0.4% DMSO) containing 0.2 µL of compounds. After 72 h, cells were washed, fixed with 4% paraformaldehyde, and stained with Hoechst 33,342 (4 µg/mL). Images (five per well) were acquired using an Operetta microscope (20× objective) and analyzed with Columbus software. Data on cell count and infection ratio were processed in GraphPad Prism 9.5.0.
RESULTS
In silico identification of putative IMPDH inhibitors in the Chagas Box collection
To find structural similarities between IMPDH inhibitors and T. cruzi active compounds in the Chagas Box, we compared the SMILES of prokaryotic IMPDH inhibitors (A110, C64, C91), or human IMPDH inhibitors (mycophenolate, mizoribine, merimepodib and AVN-944) –shown in Fig. 1A– with the Chagas Box library using Tanimoto similarity scoring (Fig. 1B). IMPDH prokaryotic inhibitors yielded no significant Tanimoto scoring with any of the Chagas Box compound set, while when using merimepodib and AVN-944, the analysis revealed that only TCMDC-143376 exhibited Tanimoto indexes of 0.817 and 0.506 for any enantiomer of merimepodib and AVN-944, respectively. Such scores were statistically different (α = 0.0001) from those displayed by the rest of the Chagas Box molecules. This result highlights the unique structural resemblance of TCMDC-143376.
Structures of IMPDH inhibitors and structural similarity analysis of Chagas Box compounds. (A) Chemical structures of the reference inhibitors used in the analysis, including their corresponding Tanimoto similarity scores relative to the compound TCMDC-143376 (phenotypic hit from Chagas Box, enclosed with a red rectangle). (B) Tanimoto similarity indices of 222 Chagas Box compounds, computed against a set of known IMPDH inhibitors: AVN-944, (S)-merimepodib, (R)-merimepodib, mycophenolate mofetil, mizoribine, and the prokaryotic inhibitors A110, C91, and C64. Iterative Grubbs’ method with an α = 0.0001 to identify outliers inside each sample was used (GraphPad Prism 9.5.0).
A phylogenetic analysis performed using IMPDH sequences from across the tree of life (see Fig. S1) revealed that IMPDHs from Trypanosomatidae cluster with those from Chordata and Fungi. This evolutionary proximity corroborates the observed in silico cross-species structure similarities of inhibitors such as AVN-944 and merimepodib. In contrast, most Apicomplexan IMPDHs form a distinct clade within eukaryotes, separate from their prokaryotic counterparts, with the notable exception of the Cryptosporidium genus, whose IMPDH sequences group within the bacterial cluster, as previously reported (28, 57).
It is likely that TCMDC-143376 may be targeting T. cruzi IMPDH. This structural insight provided a strong rationale for further investigating TcIMPDH as the potential molecular target of TCMDC-143376, and by extension, for exploring the entire Chagas Box library in the context of IMPDH inhibition. Therefore, we proceeded to study, clone, and recombinantly express Trypanosoma cruzi IMPDH to enable further biochemical and pharmacological characterization of this enzyme as a potential drug target.
Molecular and biochemical characterization of TcIMPDH
The IMPDH gene from T. cruzi was identified in the kinetoplastid genomic resource database (https://tritrypdb.org/tritrypdb/app) under the code TcCLB.507211.40 located in chromosome 8. In the T. cruzi genome, there exists another copy of this gene, located in chromosome 40 with a 98% identity, with accession code TcCLB.511351.9. The TcIMPDH gene is a 1,539-bp long open-reading frame that encodes for a polypeptide of 512 amino acids with a predicted molecular mass of 55.6 kDa. Both protein isoforms have been identified in proteomic studies of T. cruzi glycosomes, consistent with the presence of a canonical peroxisomal targeting signal type 1 (PTS1, SKL) in both sequences (21). IMPDH is a member of the Pfam00571 family, characterized by the IMPDH/GMP reductase domain, which adopts a canonical TIM barrel structure (α/β-barrel), composed of eight α-helices and eight parallel β-strands (58). In addition to the catalytic core, two CBS (cystathionine beta-synthase) domains are inserted within the barrel, often associated with allosteric regulation as found elsewhere for other IMPDHs and potentially occurring in Trypanosomatid IMPDHs, as was observed in T. brucei IMPDH CBS domain interacting with GMP and ATP in cellulo crystals (49).
Based on the structure of T. foetus IMPDH in complex with XMP and NAD^+^ (59), the binding sites of this structure were mapped in TcIMPDH. XMP/IMP site is defined by a conserved set of residues: S317, D358, E408, G409, and E431. Through alignment (see Fig. S2), we identified the equivalent residues in T. cruzi IMPDH as S323, D358, M408 (instead of E as in T. foetus), G409, and Q435 (replacing E found in T. foetus). In relation to the NAD^+^, binding residues identified in T. foetus (59) are G314, D261, S262, S263, R241, and W269. In T. cruzi, the equivalent residues are G320, D268, S269, S270, R247, and Y276. The conservation of core catalytic residues across bacteria, protozoan, and mammalian species supports a shared catalytic mechanism, also observed by other authors (60).
In addition to the conservation of catalytic residues, alignment-based comparison between T. cruzi and T. brucei IMPDH—proteins sharing 81% sequence identity—provides insight into the regulatory Bateman domains. Structural studies of T. brucei IMPDH (TbIMPDH) obtained from in cellulo crystals (49) identified a GMP binding site within this domain, involving residues K115, S136, and G137. These same residues are conserved in T. cruzi IMPDH (TcIMPDH) at identical positions (see Fig. S2), suggesting functional conservation. Furthermore, the ATP-binding site in TbIMPDH is composed of S136, T156, K157, D158, T174, T180, H200, Y202, and R219. In TcIMPDH, all these residues are conserved, with the exception of a single conservative substitution of T156 by a serine residue. This high degree of conservation reinforces the structural and functional similarities between these ortholog enzymes.
To enable further analysis of TcIMPDH, the purified recombinant enzyme was kinetically characterized. Steady-state activities were fitted to the Michaelis-Menten model to derive kinetic parameters. The resulting Km values were 155 μM (95% confidence interval 113.3–197.8 μM) and 292 μM (95% confidence interval 221.6–364.0 μM) for IMP and NAD^+^, respectively (Fig. S3). Compared with T. brucei IMPDH (Km(IMP) = 30 μM, Km(NAD^+^) = 1,300 μM) (61) and L. donovani IMPDH (Km(IMP) = 33 μM, Km(NAD^+^) = 390 μM) (62), TcIMPDH shows a moderately lower affinity for IMP but an intermediate affinity for NAD^+^. These values also contrast sharply with human IMPDHs, which exhibit much tighter binding to both substrates (Km(IMP) = 4–18 μM; Km(NAD^+^) = 6–70 μM) (63).
Screening of the Chagas Box
To validate the in silico predictions, we screened the Chagas Box collection for TcIMPDH inhibitors, which was kindly provided by GSK in a format of ready-to-use 384-well black plates (for more details, see Materials and Methods). The screening assay employed the coupled reaction system, in which TcIMPDH activity was linked to diaphorase-mediated resazurin reduction as previously described (52). The primary screening, conducted in two independent runs (Fig. 2), consistently identified a single compound as a putative TcIMPDH inhibitor: TCMDC-143376. This result corroborates our in silico prediction. This compound consistently showed 55%–60% inhibition across two independent runs, suggesting an IC_50_ below 10 μM (the fixed concentration used for all compounds in the Chagas Box screening). To validate inhibitory activity, GSK kindly provided resupplied samples, enabling further confirmatory assays.
Chagas Box screening for identification of TcIMPDH modulators. The experiment was performed in duplicate, and TcIMPDH activity in the presence of each sample was normalized using negative control reactions without addition of the IMP substrate (0% activity) and positive reactions without compounds (100% activity).
Confirming TCMDC-143376 as an IMPDH inhibitor and assessing other compounds against T. cruzi IMPDH
Half-maximal inhibitory concentrations (IC_50_) were measured to confirm the inhibition of TcIMPDH by TCMDC-143376 and assess other structurally similar compounds, including mycophenolate mofetil, AVN-944, and merimepodib enantiomers. They showed inhibitory activity against TcIMPDH, with IC_50_ values in the low micromolar to submicromolar range. TCMDC-143376 exhibited an IC_50_ of 4.00 µM, comparable to that of mycophenolate mofetil (4.40 µM), while AVN-944 and (S)-merimepodib demonstrated much stronger inhibition, with IC_50_ values of 0.20 µM and 0.21 µM, respectively. (R)-merimepodib showed slightly reduced potency with an IC_50_ of 0.37 µM (Table 1). In light of the IC_50_ measurement and assuming an uncompetitive inhibition mechanism (the most likely mechanism), an apparent inhibition constant (Ki ≃ 81 nM) was estimated for AVN-944 using the Cheng–Prusoff relationship adapted for tight-binding inhibitors (64). This value is similar to the Kis reported for human IMPDH isoenzymes (6–10 nM; [23]). Such similarity highlights the conserved druggability of IMPDH across species and the strong inhibitory potential of AVN-944 against TcIMPDH.
On the other hand, consistent with in silico results, mizoribine did not show measurable inhibition of TcIMPDH activity under the conditions tested. These results confirm the susceptibility of TcIMPDH to structurally similar IMPDH inhibitors and support the utility of AVN-944 and merimepodib enantiomers as potential hit compounds.
In vitro activity of IMPDH inhibitors against T. cruzi amastigotes into H9c2 host cells
To evaluate the therapeutic potential of IMPDH inhibitors against T. cruzi intracellular stage, we determined their half-maximal effective concentrations (EC_50_) in H9c2 rat cardiomyoblasts using an image-based infection assay as described by reference 56. Representative images are shown in Fig. 3, with close-ups highlighting amastigote clearance at comparable compound concentrations (1.2 µM).
In vitro effect of benznidazole and AVN-944 on T. cruzi amastigotes in H9c2 cells. H9c2 cardiomyoblast cells were infected with T. cruzi trypomastigotes, washed to remove extracellular parasites, and then incubated with the corresponding compound. Representative micrographs are shown for each condition (A) vehicle control (DMSO), (B) benznidazole (1.2 μM), or (C) AVN-944 (1.2 μM), accompanied by close-up images highlighting intracellular amastigotes. Panels D and E depict dose-response curves fitted to a four-parameter logistic model for benznidazole and AVN-944, respectively. The normalized infected cells (red) and host cell viability (blue) are plotted with corresponding standard deviations (N ≥ 3).
Among the tested molecules, AVN-944 exhibited the highest potency (EC_50_ = 0.40 µM), outperforming the reference drug benznidazole (EC₅₀ = 3.00 µM). Mycophenolate mofetil and merimepodib enantiomers also showed to be active against amastigotes, with EC_50_ values ranging from 1.5 to 4.8 µM. For comparative purposes, the previously reported anti-amastigote activity of TCMDC-143376 (~15.9 µM) was included, confirming its lower potency relative to the IMPDH-targeting compounds (Table 2).
Importantly, in our image-based assay, H9c2 cell population nearly doubled after 72 h, and—as previously shown by (56)—this proliferation is not affected by T. cruzi infection. This pattern is also observed for benznidazole-treated cells, since this drug does not interfere with host IMPDH activity. In contrast, cells exposed to IMPDH inhibitors display a distinct cytostatic phenotype: proliferation is arrested, but cells remain viable and retain normal morphology. This effect is well documented for mammalian cells treated with IMPDH inhibitors (38, 65–68). Consequently, during the 72-h assay, growth inhibition prevents a typical dose-response pattern in total host-cell counts that fall under 50% viability when compared to untreated controls, as illustrated for AVN-944 in Fig. 3E. Due to these assay limitations, half-maximal cytotoxic concentration (CC_50_) values could not be reliably determined.
DISCUSSION
In this study, we employed an integrated computational, biochemical, and cellular strategy to validate TcIMPDH as an alternative therapeutic target for Chagas disease. Tanimoto-based similarity screening of known phenotypic hits from GSK Chagas Box identified TCMDC-143376 as structurally related to known human IMPDH inhibitors Merimepodib and AVN-944 (44). Further in silico studies by phylogeny with IMPDH primary sequences revealed that Trypanosomatidae’s ones cluster with vertebrate and fungal orthologs, supporting previous results with Tanimoto indexing and giving insights about possible cross-species activity of these inhibitors. In light of these in silico results, we cloned, expressed, and kinetically characterized the TcIMPDH, enabling biochemical validation. Indeed, TCMDC-143376 inhibited TcIMPDH activity, consistent with our in silico approach, but showed moderate potency, comparable to mycophenolate mofetil, while AVN-944, (S)-merimepodib, and (R)-merimepodib were submicromolar inhibitors. Mizoribine was inactive, validating the computational prioritization. In infected H9c2 cardiomyoblasts, AVN-944 demonstrated the highest potency, outperforming the first-line treatment drug: benznidazole. Together, these results position IMPDH as a promising target for the development of drugs against Chagas disease, and by extension for other related diseases such as leishmaniasis and sleeping sickness.
Concretely, IMPDH inhibition targets the bottleneck of the purine salvage pathway, which emerges as a promising route for therapeutic intervention. Despite the apparent redundancy of the guanine nucleotide salvage pathway in trypanosomatids (see Fig. 4)—where GMP can be synthesized either directly from guanine via HGPRT, or indirectly from hypoxanthine through the sequential actions of HGPRT, IMPDH, and GMPS—evidence suggests that the latter route is predominantly utilized. This is supported by the essentiality of GMPS in T. brucei, as demonstrated by Li et al. (22), who found that GMPS-null parasites could not sustain growth unless provided with supraphysiological concentrations of guanine (100 µM), far exceeding those encountered in physiological environment. Moreover, hypoxanthine acted as a competitive inhibitor of guanine-mediated rescue, implying a shared uptake or metabolic channel between these purines. Since GMPS catalyzes the final conversion of XMP (the product of IMPDH) into GMP (see Fig. 4), these findings indicate that guanine uptake and conversion alone are insufficient under normal conditions to compensate for loss of the IMPDH-GMPS route (see Fig. 4). Thus, despite theoretical redundancy, the metabolic flux in trypanosomatids heavily favors GMP production via hypoxanthine salvage through IMPDH and GMPS, reinforcing the critical role of these enzymes in nucleotide homeostasis and parasite viability.
Schematic representation of purine salvage metabolism in T. cruzi showing the context of IMPDH role. IMP plays a central role, being derived for the production of AMP or GMP, recycling steps catalyzed by 5 and 8 return GMP or AMP to IMP central pool. In Trypanosomatids, despite apparent redundancy for GMP production, it is mainly synthesized through the sequential action of 1, 3, and 4 from hypoxanthine, rather than directly through one from guanine. Enzymes: 1: hypoxanthine-guanine phosphoribosyltransferase (HGPRT), 2: adenine phosphoribosyltransferase (APRT), 3: inosine-5-monophosphate dehydrogenase (IMPDH), 4: GMP synthase (GMPS), 5: GMP reductase (GMPR), 6: adenylosuccinate synthase, 7: adenylosuccinate lyase, 8: AMP deaminase. Abbreviations: PRPP: phosphoribosyl pyrophosphate, PPi: pyrophosphate, IMP: inosine-5-monophosphate, AMP: adenosine-5-monophosphate, GMP: guanosine-5-monophosphate, XMP: xanthosine-5-monophosphate, NAD+: nicotinamide adenine dinucleotide oxidized form, NADH: nicotinamide adenine dinucleotide reduced form, Gln: l-glutamine, Glu: l-glutamate, Asp: l-aspartate, Pi: orthophosphate.
AVN-944 exhibited potent antiparasitic activity in our in vitro model using H9c2 cardiomyoblasts infected with T. cruzi, significantly outperforming both benznidazole—the current frontline therapy—and mycophenolate mofetil. Specifically, AVN-944 achieved the lowest EC_50_ among the compounds tested, demonstrating the highest potency against T. cruzi amastigotes. Beyond its in vitro potency, AVN-944 is already in phase II clinical trials for oncology indications, which suggests, despite not having been published, it may have undergone extensive pharmacokinetic and toxicological evaluation in humans. These encouraging results justify further investigation of AVN-944 in animal models as a proof of concept for targeting T. cruzi IMPDH, which may be extended for other trypanosomatids too. However, given the structural similarity between AVN-944 and TCMDC-143376—previously identified as a CYP51 inhibitor with an IC_50_ ≃ 25 μM (43)—it is also possible that the trypanocidal effect of AVN-944 arises, at least in part, from CYP51 inhibition. Therefore, experimental assessment of CYP51 inhibition by AVN-944 will be essential to clarify its precise mode of action. Similarly, other evidence must be taken into account: mycophenolate mofetil has been tested in Chagas murine model showing the reduction of parasitemia, but it fails to improve survival outcomes or achieve parasitological cure (34). Furthermore, clinical evidence has raised safety concerns, as mycophenolate mofetil has been implicated in T. cruzi reactivation in immunosuppressed patients as part of the treatment to avoid rejection of heart transplantation (33).
The development of AVN-944 and merimepodib was originally aimed at targeting human IMPDH isoenzymes, which explains their effective performance in cancer (38, 69) and antiviral therapies (40, 65, 70). However, these therapeutic benefits are often accompanied by immunosuppressive effects. In humans, IMPDH is a key enzyme in the de novo synthesis of guanine nucleotides, which are essential for DNA and RNA synthesis, particularly during immune cell activation and proliferation. Upon T cell activation, both IMPDH1 and IMPDH2 isoforms are upregulated, leading to a sharp increase in IMPDH activity that supports the rapid expansion required for an effective immune response (71–73). This immunological context is highly relevant for Chagas disease, where a robust immune response is crucial. During the acute phase, both B and T lymphocytes undergo marked expansion (74, 75). The balance between effector and regulatory lymphocytes is also critical. For instance, regulatory T cells (Tregs) expand in individuals with the indeterminate (asymptomatic) form of Chagas disease, helping to limit tissue damage by controlling hyperactive immune responses (76). Similarly, expansion of specific B cell subsets—such as CD11b^+^ B1 B cells—has been associated with improved cardiac function and a protective immune profile (77, 78). And finally, in the chronic phase, CD4^+^CD8^+^ double-positive T cells retain the ability to produce effector molecules against the parasite (79).
The limitations of current IMPDH inhibitors—particularly their immunosuppressive effects—highlight the need for more selective compounds. Although there is evidence that AVN-944 and merimepodib are likely acting as uncompetitive inhibitors of IMPDH (23), their exact binding site on the enzyme remains unidentified. Comparison of the estimated Ki values for AVN-944 between TcIMPDH and the human orthologs indicates greater potency toward the human IMPDH isoenzymes than the parasite counterpart, underscoring the need to improve selectivity for the T. cruzi target over human IMPDHs. Determining the precise binding site is crucial, as it would enable the rational design of new inhibitors that specifically target trypanosomatid IMPDHs while sparing the human isoforms. This selectivity is especially important not only to avoid side effects but also to preserve the host’s natural immune response, which plays a critical role in controlling T. cruzi infection and improving clinical outcomes (71–79). While some structural insights have been gained from the recently solved in cellulo crystal structure of T. brucei IMPDH (49), the binding pocket for these inhibitors has not yet been mapped. Identifying this region will be key to exploiting structural differences between human and parasite enzymes, and it will guide the repurposing of AVN-944 and Merimepodib scaffold for the development of safer, more effective chemotherapies that work in synergy with the host immune system.
Conclusions
This study identifies TcIMPDH as a promising drug target for Chagas disease through an integrated in silico, biochemical, and in vitro cellular approach. Using ligand-based virtual screening, we linked the Chagas Box hit TCMDC-143376 to known human IMPDH inhibitors, notably AVN-944 and merimepodib. Biochemical and kinetic validation confirmed TcIMPDH inhibition, with AVN-944 displaying potent enzymatic and antiparasitic activity, outperforming benznidazole in infected cardiomyoblasts. Our results support the essentiality of guanine nucleotide salvaging via IMPDH in T. cruzi, despite the theoretical redundancy of the purine salvage pathway. Importantly, AVN-944’s submicromolar efficacy and clinical-stage status underscore its potential to perform the proof of concept of targeting IMPDH in animal models. However, its known immunosuppressive effects in humans highlight the need for more selective inhibitors that preserve host immune function. Mapping the binding site of AVN-944 on trypanosomatid IMPDH will be critical for the rational design of parasite-selective compounds with improved safety and efficacy profiles for Chagas disease and related kinetoplastid infections.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lidani KCF, Andrade FA, Bavia L, Damasceno FS, Beltrame MH, Messias-Reason IJ, Sandri TL. 2019. Chagas disease: from discovery to a worldwide health problem. Front Public Health 7:166. doi:10.3389/fpubh.2019.0016631312626 PMC 6614205 · doi ↗ · pubmed ↗
- 2Conners EE, Vinetz JM, Weeks JR, Brouwer KC. 2016. A global systematic review of Chagas disease prevalence among migrants. Acta Trop 156:68–78. doi:10.1016/j.actatropica.2016.01.00226777312 PMC 5155706 · doi ↗ · pubmed ↗
- 3Cucunubá ZM, Gutiérrez-Romero SA, Ramírez J-D, Velásquez-Ortiz N, Ceccarelli S, Parra-Henao G, Henao-Martínez AF, Rabinovich J, Basáñez M-G, Nouvellet P, Abad-Franch F. 2024. The epidemiology of Chagas disease in the Americas. The Lancet Regional Health - Americas 37:100881. doi:10.1016/j.lana.2024.10088139474465 PMC 11519694 · doi ↗ · pubmed ↗
- 4García-Huertas P, Cardona-Castro N. 2021. Advances in the treatment of Chagas disease: promising new drugs, plants and targets. Biomed Pharmacother 142:112020. doi:10.1016/j.biopha.2021.11202034392087 · doi ↗ · pubmed ↗
- 5Jayawardhana S, Ward AI, Francisco AF, Lewis MD, Taylor MC, Kelly JM, Olmo F. 2023. Benznidazole treatment leads to DNA damage in Trypanosoma cruzi and the persistence of rare widely dispersed non-replicative amastigotes in mice. PLOS Pathog 19:e 1011627. doi:10.1371/journal.ppat.101162737956215 PMC 10681306 · doi ↗ · pubmed ↗
- 6Mejia AM, Hall BS, Taylor MC, Gómez-Palacio A, Wilkinson SR, Triana-Chávez O, Kelly JM. 2012. Benznidazole-resistance in Trypanosoma cruzi is a readily acquired trait that can arise independently in a single population. J Infect Dis 206:220–228. doi:10.1093/infdis/jis 33122551809 PMC 3379838 · doi ↗ · pubmed ↗
- 7Hall BS, Bot C, Wilkinson SR. 2011. Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J Biol Chem 286:13088–13095. doi:10.1074/jbc.M 111.23084721345801 PMC 3075655 · doi ↗ · pubmed ↗
- 8Lepesheva GI, Villalta F, Waterman MR. 2011. Targeting Trypanosoma cruzi Sterol 620 14α-Demethylase (CYP 51), p 65–87. In Advances in Parasitology. Vol. 75. Elsevier.21820552 10.1016/B 978-0-12-385863-4.00004-6PMC 3488290 · doi ↗ · pubmed ↗