Emergence and genomic adaptation of the globally disseminated ST2250 lineage within the Staphylococcus aureus complex
Wangxiao Zhou, Dizhong Chen, Xu Dong, Ting Yang, Caixia Liu, Ye Jin, Deru Lei

TL;DR
This study explores the global spread and genomic evolution of the ST2250 lineage of Staphylococcus argenteus, revealing its adaptation and resistance mechanisms.
Contribution
The study provides new insights into the evolutionary history and genomic adaptations of the ST2250 lineage of S. argenteus.
Findings
ST2250 lineage diversified into three clades with cross-regional transmission events.
A methicillin-resistant subclade in clade II arose from a single SCCmec IVc acquisition.
Clade III carried a multidrug-resistant plasmid introduced around 2001, aiding its expansion.
Abstract
Staphylococcus argenteus, a member of the S. aureus complex, is increasingly recognized as a globally distributed pathogen with significant clinical relevance. Among its lineages, sequence type (ST) 2250 has emerged as the most prevalent and geographically widespread, yet its evolutionary history and genomic adaptations remain incompletely understood. In this study, we conducted a comprehensive genomic analysis of 277 ST2250 genomes from 26 countries between 2008 and 2025, integrating 14 newly sequenced isolates from China. Phylogenetic reconstruction resolved a basal clade I around 1989 and sister clades II and III that diversified later, in approximately 1996 and 1997, with frequent cross-regional, intercontinental, and cross-host transmission events. A methicillin-resistant S. argenteus subclade within clade II likely arose from a single SCCmec IVc acquisition, accompanied by a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
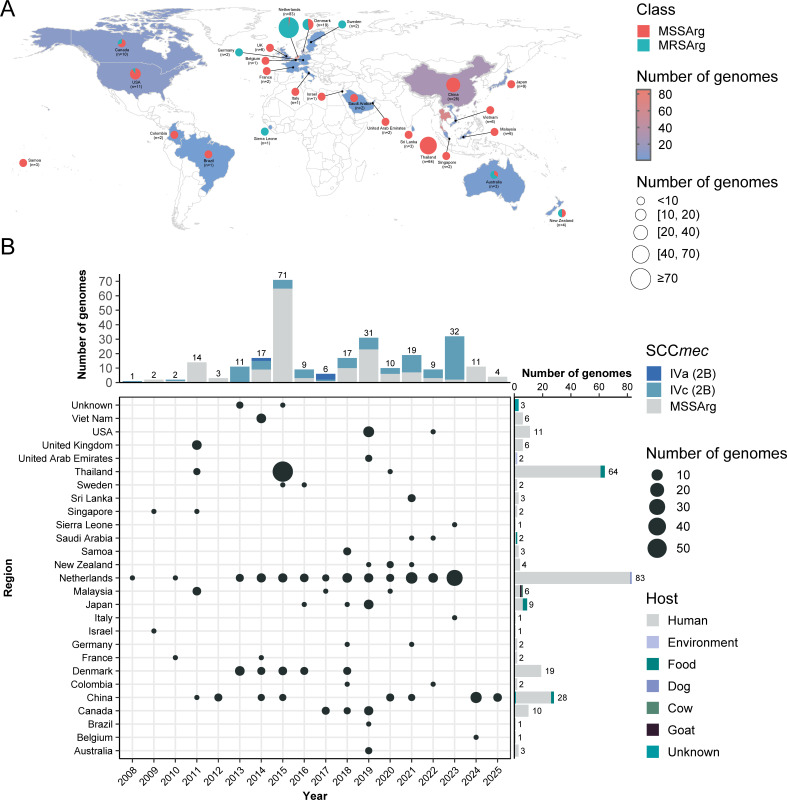 Fig 1
Fig 1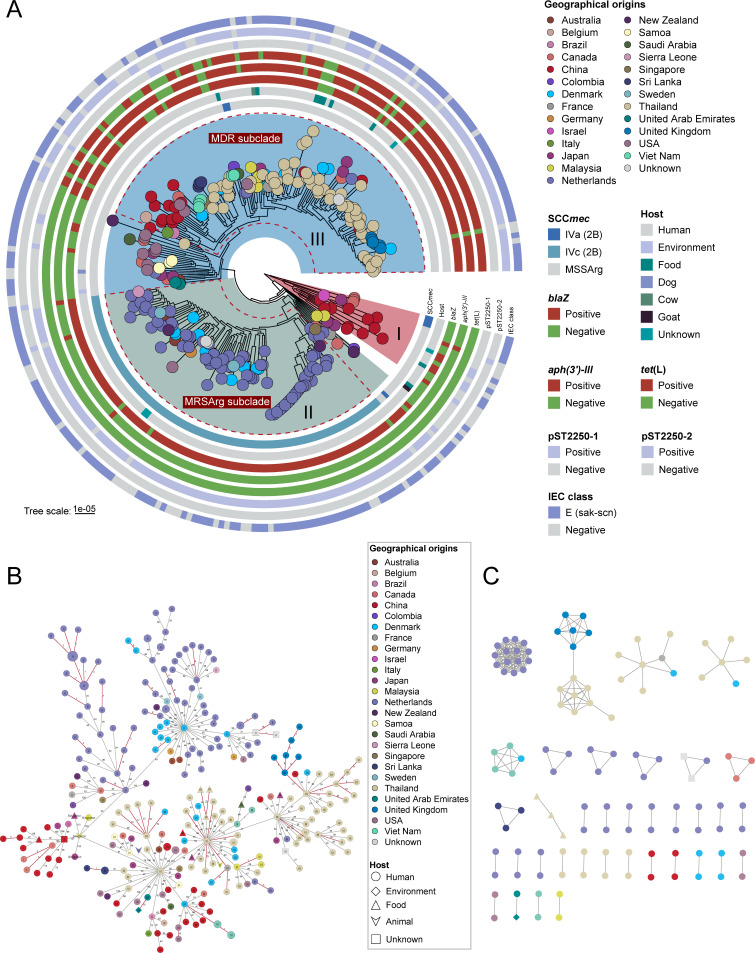 Fig 2
Fig 2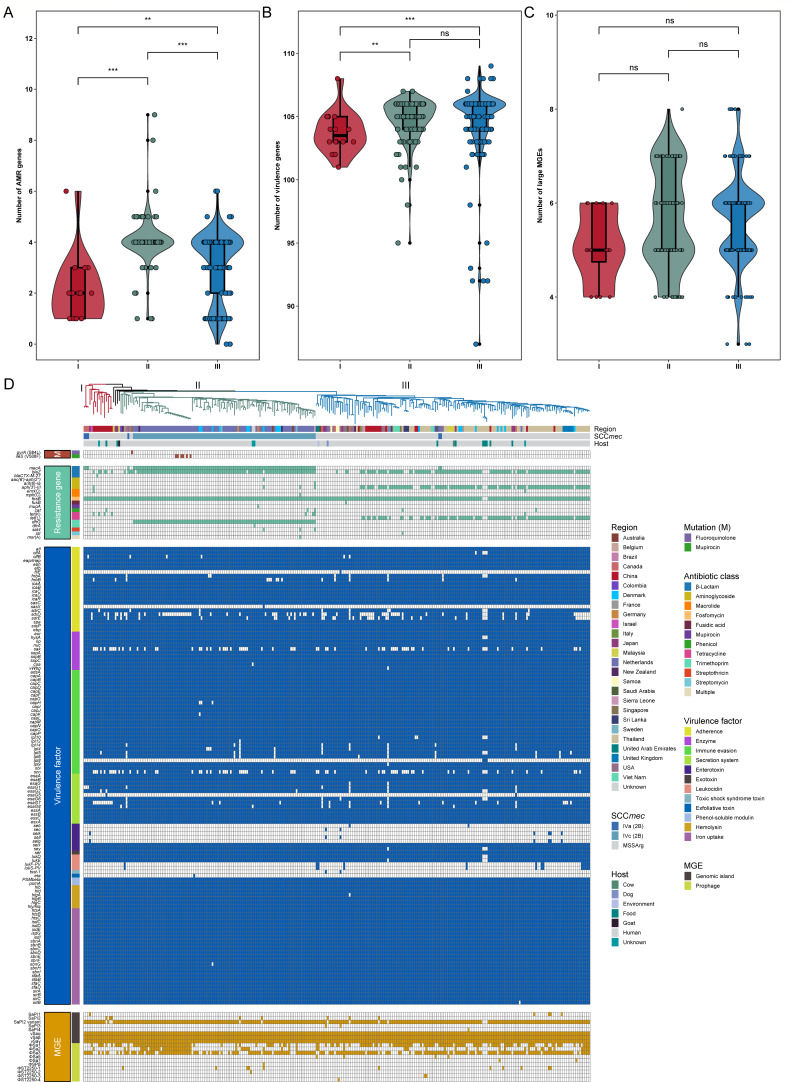 Fig 3
Fig 3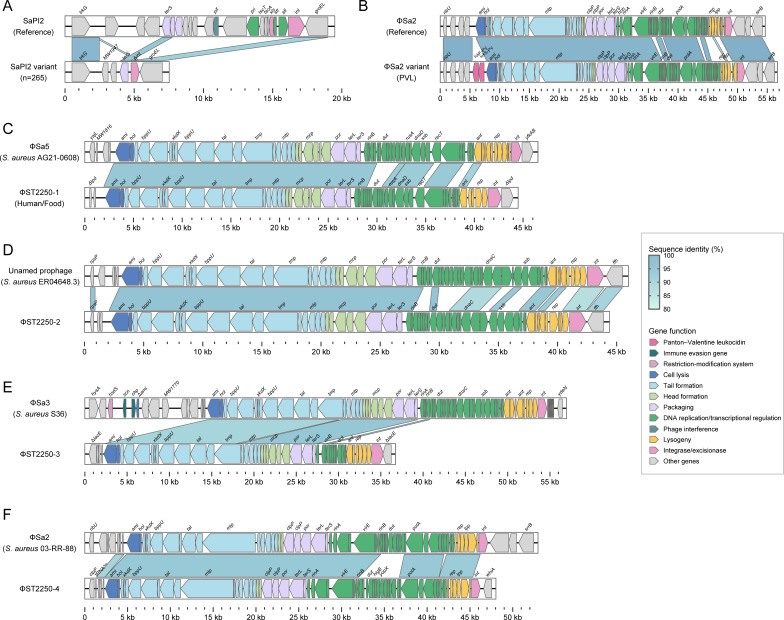 Fig 4
Fig 4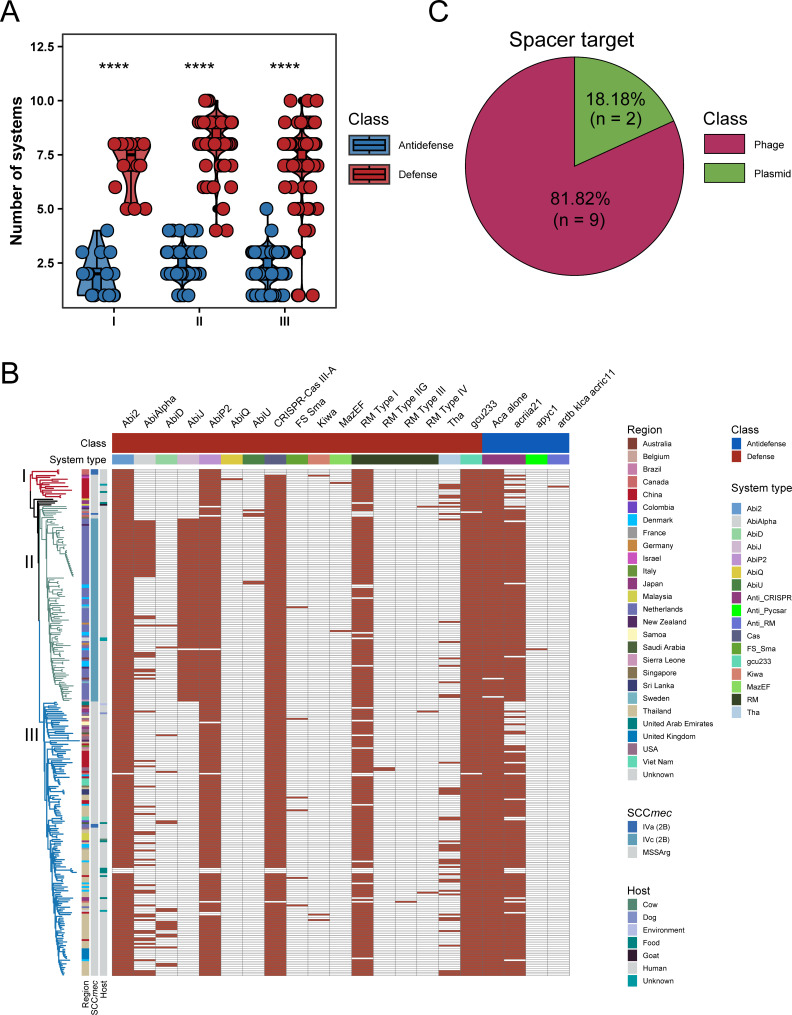 Fig 5
Fig 5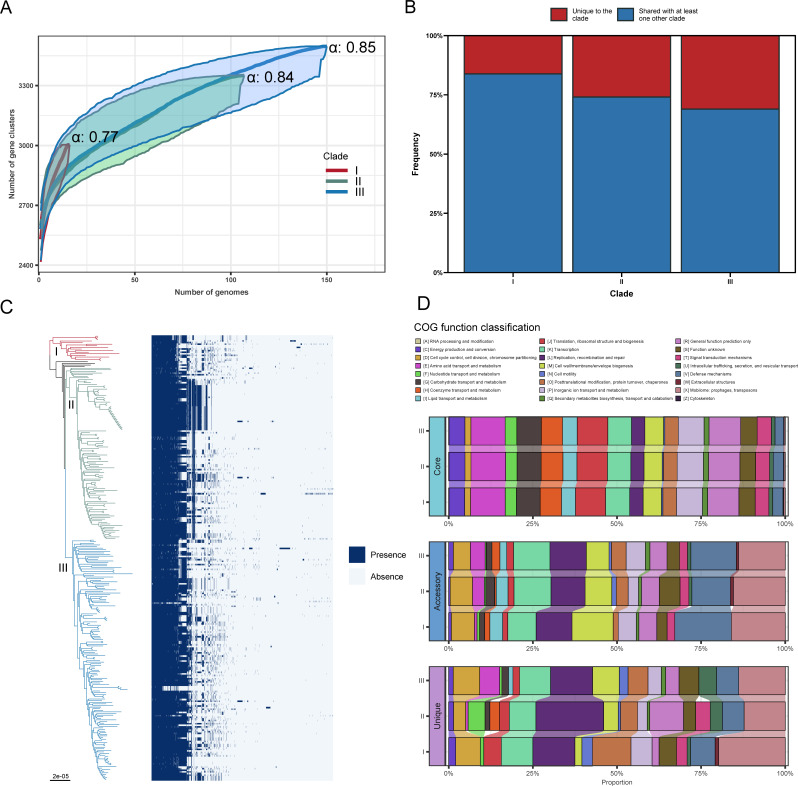 Fig 6
Fig 6| Isolate | Isolation date | ST | SCC | Age (year) | Gender | Specimen | Ward | Primary disease | Outcome | |
|---|---|---|---|---|---|---|---|---|---|---|
| FEYWZ100 | 2024-11-27 | ST2250_1LV | MSSArg | t5078 | 81 | Male | Respiratory tract | Critical care medicine | Pneumonia | Discharged |
| FEYWZ123 | 2024-12-06 | ST2250 | MSSArg | t7960 | 16 | Male | Respiratory tract | Emergency medicine | Pneumonia | Discharged |
| FEYWZ132 | 2024-12-11 | ST2250 | MSSArg | t6675 | 56 | Female | Respiratory tract | Integrated rehabilitation | Pneumonia | Discharged |
| FEYWZ175 | 2024-12-30 | ST2250 | MSSArg | Undetermined | 29 | Male | Respiratory tract | Emergency medicine | Cerebral hemorrhage | Discharged |
| FEYWZ217 | 2025-01-16 | ST2250 | MSSArg | t5078 | 53 | Male | Wound/pus | Hand surgery | Osteomyelitis | Discharged |
| FEYWZ357 | 2025-03-10 | ST2250 | MSSArg | Undetermined | 89 | Male | Blood | Emergency medicine | Pneumonia | Dead |
| FEYWZ35 | 2024-11-04 | ST2250 | MSSArg | t5078 | 64 | Female | Urine | Urology surgery | Urinary tract infection | Discharged |
| FEYWZ369 | 2025-03-19 | ST2250 | MSSArg | Undetermined | 34 | Male | Wound/pus | Colorectal and anal surgery | Perihepatic abscess | Discharged |
| FEYWZ372 | 2025-03-21 | ST2250 | MSSArg | t5078 | 55 | Male | Others | Hepato-pancreato-biliary surgery | Chronic cholecystitis | Discharged |
| FEYWZ6 | 2024-10-25 | ST2854 | MSSArg | Undetermined | 80 | Male | Blood | Neurosurgery | Cerebral hemorrhage | Discharged |
| FEYWZ12 | 2024-10-20 | ST2250 | MSSArg | t5078 | 83 | Male | Blood | Critical care medicine | Pneumonia | Dead |
| FEYWZ14 | 2024-10-23 | ST2250 | MSSArg | t6675 | 76 | Female | Respiratory tract | Neurology | Cerebral infarction | Discharged |
| FEYWZ15 | 2024-10-28 | ST2250 | MSSArg | t5078 | 53 | Female | Blood | Hematology | Sepsis | Dead |
| FEYWZ17 | 2024-10-29 | ST2250 | MSSArg | t5078 | 74 | Male | Blood | Infectious diseases | Sepsis | Dead |
| FEYWZ55 | 2024-11-12 | ST2250 | MSSArg | t7960 | 60 | Male | Blood | Nephrology | Obstructive nephropathy | Discharged |
| FEYWZ65 | 2024-11-15 | ST1223 | IVb (2B) | Undetermined | 41 | Female | Blood | Emergency medicine | Sepsis | Dead |
| FEYWZ82 | 2024-11-21 | ST2250 | MSSArg | t5078 | 49 | Male | Blood | Emergency medicine | Sepsis | Discharged |
| Isolate | OXA | PEN | ERY | CLI | RIF | TCY | TGC | CIP | LVX | MFX | GEN | AMK | SXT | LNZ | Q/D | VAN | DAP | BPR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FEYWZ100 | S | R | S | S | S | R | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ123 | S | R | R | R | S | R | S | S | S | S | S | R | S | S | S | S | S | S |
| FEYWZ132 | S | R | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ175 | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ217 | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ357 | S | R | S | S | S | R | S | S | S | S | S | R | S | S | S | S | S | S |
| FEYWZ35 | S | R | S | S | S | S | S | S | S | S | S | R | S | S | S | S | S | S |
| FEYWZ369 | S | R | S | S | S | R | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ372 | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ6 | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ12 | S | R | S | S | S | R | S | S | S | S | S | R | S | S | S | S | S | S |
| FEYWZ14 | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ15 | S | R | S | S | S | R | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ17 | S | R | S | S | S | S | S | S | S | S | S | R | S | S | S | S | S | S |
| FEYWZ55 | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ65 | R | R | S | S | S | R | S | S | S | S | S | S | S | S | S | S | S | S |
| FEYWZ82 | S | R | S | S | S | R | S | S | S | S | S | R | S | S | S | S | S | S |
- —Wenzhou Municipal Science and Technology Bureauhttp://dx.doi.org/10.13039/501100007194
- —National Natural Science Foundation of China-Yunnan Joint Fundhttp://dx.doi.org/10.13039/501100011002
- —Zhejiang Provincial Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Resistance in Staphylococcus · Bacterial biofilms and quorum sensing · Biochemical and Structural Characterization
INTRODUCTION
Staphylococcus argenteus is a recently delineated member of the S. aureus complex (SAC), which also includes S. aureus, S. schweitzeri, S. roterodami, and S. singaporensis (1, 2). Initially recognized in 2006 during molecular epidemiological surveillance of methicillin-resistant S. aureus (MRSA) in northern Australia, S. argenteus was originally classified as a clonal complex 75 of S. aureus due to their close phenotypic similarity (3). However, subsequent whole-genome sequencing (WGS) analyses revealed substantial genetic divergence, leading to its formal designation as a separate species in 2015 (4). Since its recognition, reports of S. argenteus infection have emerged across diverse regions including Oceania, Asia, Europe, Africa, and the Americas (5–8). In clinical isolates from Asia, the proportion of S. argenteus typically remains below 10% among presumptive S. aureus isolates (5, 6, 9). Nevertheless, comprehensive epidemiological data from China remain limited, and the clinical relevance of S. argenteus infections in this region is still not fully understood. A few recent studies have detected S. argenteus in clinical infections and retail food samples in southern China (10, 11), raising public health concerns regarding potential foodborne or zoonotic transmission routes.
Although S. argenteus was initially considered less virulent than S. aureus, largely due to the absence of staphyloxanthin (12), accumulating evidence now indicates that it can cause a broad spectrum of severe infections, including osteomyelitis, prosthetic joint infections, infective endocarditis, mycotic aneurysm, and bloodstream infections (13). In fact, more recent clinical data show that patients with S. argenteus bacteremia experience higher mortality rates than those infected with methicillin-susceptible S. aureus (MSSA) (14). Experimental studies have demonstrated that S. argenteus and S. schweitzeri exhibit significantly greater cytotoxicity toward human cells, which has been linked to a 12- to 15-fold increase in the expression of α-hemolysin compared with S. aureus (15). A comparative genomic study has shown that approximately 76% of the 111 virulence-associated genes typically found in S. aureus are also present in S. argenteus (16). Beyond virulence, S. argenteus can exhibit various resistance due to the presence of genes such as blaZ, fusA, and tet(L) (17), underscoring its growing potential as both a pathogen and a reservoir for antimicrobial resistance.
Among the currently recognized lineages of S. argenteus, sequence type (ST) 2250 has emerged as the most dominant and globally disseminated clone in recent years (17). In Thailand, where it has been most extensively studied, ST2250 isolates are typically susceptible to methicillin but resistant to penicillin (18). By contrast, isolates from Australia and the UK frequently harbor staphylococcal cassette chromosome mec (SCCmec) type IV, conferring methicillin resistance (19). However, these observed patterns could be influenced by biased isolate selection and may not accurately reflect regional resistance trends. Beyond resistance, ST2250 has also been implicated in persistent infections, including cases of prosthetic joint infection where isolates demonstrated small-colony variant formation and increased intracellular persistence that may be further induced under antibiotic exposure (20). Such features may facilitate chronic infection and complicate treatment. Despite its prominence, most previous research has focused on S. argenteus at the species level, leaving lineage-specific traits underexplored. A focused investigation of the ST2250 genome is needed to determine whether this lineage possesses unique evolutionary, resistance, or virulence characteristics that contribute to its apparent success.
To elucidate the evolutionary mechanisms and genomic diversity of S. argenteus ST2250, we conducted a large-scale comparative genomic analysis of 277 genomes, including newly sequenced local clinical isolates and publicly available global data. Through phylogenetic reconstruction and molecular evolutionary analyses, we delineated its population structure, geographic dissemination, and patterns of adaptive evolution. We further examined the distribution of antimicrobial resistance (AMR) genes, virulence factors, mobile genetic elements (MGEs), defense systems, and genes under positive selection to investigate the genomic basis of its clinical success and ecological adaptability. This study provides a foundational resource for understanding the evolutionary trajectory and genetic makeup of ST2250, contributing essential insights for combating S. argenteus infections.
MATERIALS AND METHODS
Collection of bacterial isolates
Between October 2024 and April 2025, a total of 435 consecutive, non-duplicate presumptive S. aureus complex isolates were prospectively collected from clinical specimens submitted for routine diagnostic testing at the Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University (Zhejiang Province, China). Duplicate isolates were defined as multiple isolates of the same species from the same patient during the study period, and only the first isolate per patient was retained. No additional selection criteria were applied. Initial species identification was performed using MALDI-TOF MS (Bruker, Bremen, Germany). Antimicrobial susceptibility testing (AST) was conducted using the VITEK-2 automated system (bioMérieux, France) following the manufacturer’s protocols. In addition, AST for daptomycin and ceftobiprole was performed by broth microdilution in accordance with CLSI guidelines.
Whole-genome sequencing and genomic analysis
Whole-genome sequencing of all 435 presumptive S. aureus complex isolates was carried out using the Illumina HiSeq X Ten platform (Illumina, San Diego, CA, USA) with 2 × 150 bp paired-end reads. Raw sequencing data were processed using fastp v0.23.2 (21) for adapter trimming and quality filtering (minimum Phred score ≥20), followed by de novo assembly with SPAdes v3.14.1 (22). Species-level classification was performed using GTDB-Tk v2.4.0 (23), which identified 17 isolates as S. argenteus; the remaining isolates were confirmed as S. aureus. In silico multi-locus sequence typing (MLST) of S. argenteus was performed using the MLST 2.0 webserver (https://cge.food.dtu.dk/services/MLST/), which revealed that 14 of the 17 S. argenteus isolates belonged to the ST2250 lineage. These ST2250 isolates were selected for downstream comparative analyses. Additionally, long-read sequencing of a representative ST2250 isolate (strain FEYWZ357) was conducted on the Oxford Nanopore MinION platform, and hybrid assembly of the complete genome was achieved by integrating Illumina short-read and Nanopore long-read data using Unicycler v0.5.0 (24).
To expand the data set, 450 publicly available S. argenteus genomes were downloaded from the NCBI genome database (as of July 2024). In silico MLST was conducted on these genomes, and only ST2250 genomes were retained for further phylogenomic analysis, resulting in a final data set of 277 ST2250 genomes (Table S1).
In silico typing of SCCmec elements and spa types was carried out using SCCion (https://github.com/esteinig/sccion). Genome annotation was performed using Bakta v1.11.2 (25), and pan-genome analysis of ST2250 genomes was conducted in strict mode using Panaroo v1.3.0 (26). The evolutionary dynamics of gene exchange within the ST2250 lineage were assessed using Panstripe v0.3.0 (27), applying a generalized linear model with a Tweedie distribution. The bacterial defense and anti-defense systems were examined across all ST2250 genomes using DefenseFinder v2.0.1 (https://github.com/mdmparis/defense-finder).
Phylogenetic analysis of ST2250 lineage
Alignment of 277 ST2250 genomes against the reference genome S. argenteus TWCC 58113 (GenBank accession no. AP018562.1) was performed using Snippy v4.6.0 (https://github.com/tseemann/snippy) to extract core-genome SNPs. Gubbins v2.4.1 (28) was used to identify regions undergoing recombination within the core genome, and the resulting recombination-free core-genome SNPs were employed to construct the phylogenetic tree. Maximum-likelihood phylogenies were inferred using RAxML v8.2.12 (29) under the GTRGAMMAIX model with 1,000 bootstrap replicates. The genome of S. argenteus MSHR1132 (ST1850, GenBank: FR821777.2) was used as the outgroup. The dated phylogeny based on Bayesian inference was constructed using BactDating v1.1 (30) under a mixed model with 100 million cycles. Convergence of the model was confirmed by ensuring effective sample sizes >200 for all parameters. Geographic origin was inferred using the Bayesian Binary MCMC method implemented in RASP v4.2 (31), employing 10 parallel chains, each run for 50 million generations. A minimum spanning tree (MST) of the 277 genomes was constructed using PHYLOViZ v2.0 (https://online.phyloviz.net/index) to visualize pairwise genetic relationships.
Comparison of AMR genes, virulence genes, and MGEs among ST2250 lineage
AMR genes and virulence factors were identified using ABRicate v1.0.0 (https://github.com/tseemann/abricate) based on ResFinder and VFDB databases with default settings. Putative pathogenicity islands and prophage elements were identified using BLASTN (https://blast.ncbi.nlm.nih.gov/Blast.cgi), with published S. aureus MGE sequences as references and thresholds set at ≥85% nucleotide identity and query coverage.
In addition, all available S. aureus genome sequences (n = 85,893 as of July 2024) were downloaded from the NCBI GenBank database. STs were assigned and genomes were annotated as described above for S. argenteus. We used Assembly Dereplicator v0.3.1 (Mash distance threshold 0.001; https://github.com/rrwick/Assembly-Dereplicator) to cluster and dereplicate global ST59, ST8, and ST239 genomes, selecting 206 ST59, 241 ST8, and 136 ST239 representatives for comparison with ST2250. Methods for identifying AMR genes, virulence genes, and MGEs were the same as those applied to ST2250.
Positive selection analysis
Ancestral state reconstruction of all core genome SNPs within ST2250 lineage was performed using PAML 4 (32), and each SNP was subsequently categorized as intergenic, synonymous, or nonsynonymous based on its annotation and inferred ancestral state. Nonsynonymous to synonymous substitution (dN/dS) ratios were calculated using the NY98 model, which incorporates both codon usage and transition/transversion rate biases. Genes were considered to be under positive selection if the adjusted dN/dS ratio exceeded 1.5, or if no synonymous mutations were detected but at least seven nonsynonymous mutations were present.
Genetic origin analysis
For origin analysis of clonal complex (CC) 2250 (denoting ST2250 and its single locus variants), we computed Mash distances v2.3 (33) between TWCC 58113 and all available S. aureus (n = 85,893) and non-ST2250 S. argenteus (n = 173) genomes, selected the 500 closest genomes together with 277 ST2250 genomes for downstream phylogeny, and inferred recombination events with Gubbins v2.4.1 (28) as described above for ST2250.
Statistical analysis
Statistical analysis was conducted using the Wilcoxon rank-sum test for continuous variables and the chi-square or Fisher’s exact test for categorical variables, with P values <0.05 considered statistically significant. All data were processed and analyzed using R software (version 4.5.1).
RESULTS
Identification and prevalence of S. argenteus isolates
Between October 2024 and April 2025, a total of 435 consecutive, non-duplicate S. aureus complex isolates were obtained from a large tertiary teaching hospital in Zhejiang Province, China. Species identification by MALDI-TOF MS followed by WGS revealed that 17 isolates (3.91%) were S. argenteus. These isolates were recovered from various clinical specimens, including blood (n = 8), respiratory tract samples (n = 5), wound/pus exudates (n = 2), and one specimen each from the urinary tract and bile. All patients received antimicrobial therapy, but five (29.41%) died during hospitalization from S. argenteus infection, including three with sepsis and two with pneumonia. The clinical information of these patients is provided in Table 1.
In silico MLST revealed that the majority of the S. argenteus isolates belonged to ST2250 (82.35%, 14/17), while the remaining three isolates were classified as ST1223, ST2854, and a single-locus variant of ST2250. Among the 14 ST2250 isolates, the most common spa type was t5078 (50%, 7/14), followed by t6675 (n = 2), t7960 (n = 2), and three unassigned strains. Notably, pairwise SNP distances among ST2250 isolates ranged from 108 to 290, well above the ≤24 SNP threshold commonly used to infer recent clonal transmission in S. aureus (34), indicating no evidence of recent direct transmission in our cohort and supporting independent introductions. AST showed that all S. argenteus isolates were susceptible to vancomycin, linezolid, tigecycline, quinupristin-dalfopristin, fluoroquinolones, rifampin, trimethoprim-sulfamethoxazole, daptomycin, and ceftobiprole. However, resistance was observed in 64.71%, 47.06%, and 35.29% of isolates to penicillin, tetracycline, and amikacin, respectively. One methicillin-resistant isolate (FEYWZ65), assigned to ST1223, was also identified (Table 2). Given the predominance of the ST2250 lineage in both this study and global epidemiology (35), ST2250 isolates were selected for subsequent genomic analyses.
Geographical dissemination and phylogenomic diversity of the ST2250 lineage
To investigate the global prevalence of ST2250 lineage, all publicly available genomes labeled S. argenteus were retrieved from the NCBI GenBank database. Among 450 genomes screened, 263 were identified as ST2250 based on in silico MLST analysis. Combined with 14 ST2250 isolates from our collection, a data set comprising 277 high-quality ST2250 genomes was constructed for downstream analyses. These genomes were sampled between 2008 and 2025 from 26 countries across six continents (Fig. 1), with the highest numbers reported from the Netherlands (29.96%, 83/277), Thailand (23.10%, 64/277), and China (10.11%, 28/277). Notably, 106 of the 277 genomes (38.27%) were identified as methicillin-resistant S. argenteus (MRSArg), originating from 10 countries across Europe, North America, and Oceania. MRSArg genomes were detected nearly every year since 2008 (Fig. 1), suggesting a sustained pattern of geographic diversity and dissemination. In addition, although most genomes were sampled from human clinical samples, 13 originated from non-human sources, including food (n = 9), hospital environment (n = 1), cow (n = 1), dog (n = 1), and goat (n = 1); the source of four genomes was not reported.
Global distribution and temporal dynamics of S. argenteus ST2250 genomes. (A) Geographic location of 277 S. argenteus ST2250 genomes collected from 26 countries. Red and cyan circles represent methicillin-susceptible (MSSArg) and methicillin-resistant (MRSArg), respectively. Circle size corresponds to the number of genomes from each country, and background shading indicates regions with ST2250 detections. (B) Temporal and geographic overview of ST2250 genomes. The central bubble plot displays the number of genomes (bubble size) reported per country each year. The upper bar chart shows the annual counts of genomes by SCCmec type. The right-side bar chart summarizes the host distribution for each country.
Maximum-likelihood phylogenetic analysis based on core-genome SNPs resolved three clades (I to III; Fig. 2A) within the 277 ST2250 genomes. The time to the most recent common ancestor was estimated at 1986 (95% highest posterior density [HPD]: 1981–1990; Fig. S1). Clade I branched near the root, emerging shortly after the ST2250 ancestor around 1989, whereas clades II and III formed a sister group that diversified slightly later in the mid to late 1990s, at approximately 1996 and 1997, respectively. These findings indicate that the global spread of ST2250 has persisted for more than three decades. After excluding recombinant regions, the estimated mean nucleotide substitution rate for the ST2250 lineage was 1.58 × 10^−6^ substitutions/site/year (95% HPD: 1.41 × 10^−6^ to 1.75 × 10^−6^), comparable to that of other prevalent S. aureus lineages (36). Phylogeographic analysis indicated that clade I genomes originated from only four countries, predominantly China (68.75%, 11/16), whereas clades II and III exhibited greater geographic diversity. Notably, clade II was dominated by genomes from the Netherlands (73.83%, 79/107), while clade III included a high proportion of genomes from Thailand (42.67%, 64/150) and China (11.33%, 17/150). Additionally, aside from sporadic MRSArg carrying SCCmec IVa (n = 6), all MRSArg harboring SCCmec IVc were clustered in clade II, forming a distinct MRSArg subclade, likely resulting from a single acquisition event of SCCmec IVc around 2001.
Phylogenetic and transmission analyses of 277 S. argenteus ST2250 genomes. (A) Phylogenetic structure of ST2250 genomes. Clade I is basal, and clades II and III share a recent common ancestor. Tip colors represent the country of isolation, and the concentric rings (inner to outer) indicate SCCmec types, host types, presence/absence of resistance genes [blaZ, aph(3′)-III and tet(L)], plasmids (pST2250-1 and pST2250-2), and immune evasion cluster (IEC) gene classes. MRSArg and MDR subclades are highlighted. (B) MST of all ST2250 genomes. Each node represents one genome, with node color denoting geographic origin and shape indicating host source. Red edges denote potential transmission clusters, defined by a <24 SNP threshold. (C) Transmission clusters based on pairwise SNP distances <24. Each node represents one genome, colored by geographic origin and shaped by host source.
In addition, the paired SNP differences of genomes between three clades varied from 96 to 321 (median = 160), indicating considerable genetic divergence at the clade level. However, genomes originating from geographically distant regions, including multiple countries and continents, were tightly clustered within clades II and III (Fig. 2A and B), suggesting potential cross-country transmission. Phylogenetic analysis also showed that non-human genomes were scattered across the phylogeny and clustered with proximal human-derived genomes, indicating potential cross-host transmission. Based on a ≤24 SNP threshold, we identified 36 independent transmission events across 12 countries (Fig. 2C), including intercontinental links between Thailand and the United Kingdom, Thailand and Denmark, and Vietnam and Denmark, as well as a potential transmission event involving a patient and the clinical environment in the United Arab Emirates (37). Notably, Bayesian phylogeographic analysis supports a Japanese origin for clade I Chinese genomes and implicates a basal Singapore methicillin-susceptible S. argenteus (MSSArg) as a potential ancestor of the Dutch ST2250 MRSArg expansion, subject to sampling bias (Fig. S2).
To probe the origin of CC2250 (denoting ST2250 and its single locus variants), we reconstructed a phylogeny including the 500 closest hits to the ST2250 reference TWCC 58113 together with 277 ST2250 genomes (Fig. S3A). Within CC2250, the median pairwise core SNP distance was 131 (range 0 to 312). The closest external lineage was CC75, with a median distance of 11,126 SNPs (range 11,071 to 11,208) from CC2250 (Fig. S3B), far exceeding within CC diversity. Recombination mapping along the TWCC 58113 reference showed no single mosaic block indicative of wholesale chromosomal replacement between CC2250 and CC75; inferred recombination events were dispersed across the chromosome (Fig. S3C).
Clade-specific distribution of resistance genes among ST2250 lineage
Given the critical role of multidrug resistance in the persistence of S. argenteus in healthcare settings, we compared the resistance gene profiles across major clades within the ST2250 lineage. A total of 22 resistance genes were identified, covering 12 classes of antimicrobials. Notably, the mean number of resistance genes per genome was higher in clades II (4.05) and III (3.12) than in clade I (2.12) (P < 0.001) (Fig. 3A). Specifically, the high prevalence of MRSArg in clade II (94.39%) was associated with frequent carriage of the β-lactam resistance gene mecA (94.39%) and the trimethoprim resistance gene dfrG (93.46%), both located within the SCCmec IVc element; and the blaZ gene encoding β-lactamase was detected in 94.39% of clade II genomes, significantly higher than in clade I. In addition, clade III genomes showed a high prevalence of blaZ (74.67%), the tetracycline resistance gene tet(L) (63.33%), and the aminoglycoside resistance gene aph(3')-III (70.67%), which were enriched within a distinct multidrug-resistant (MDR) subclade (Fig. 2A and 3D).
*Distribution of AMR genes, virulence factors, and large MGEs in ST2250 genomes. (A–C) Violin plots comparing the number of AMR genes (A), virulence factors (B), and large MGEs (C) among the three ST2250 clades. **, P < 0.01; **, P < 0.001. ns, not significant. (D) Heatmap illustrating the distribution of mutations, AMR genes, virulence factors, and MGEs across ST2250 lineages. Squares colored by trait category indicate the presence of each examined trait.
Further analysis revealed that blaZ in clade II was located on a multi-replicon plasmid (rep5a/rep16), designated pST2250-1. This plasmid, approximately 20.6 kb in size, also harbored the heavy metal resistance genes cadD and arsR (Fig. S4A). BLAST search results indicated that pST2250-1 shared 100% nucleotide identity with S. aureus plasmid pMW2 (AP004832.1), which was originally identified in a human clinical isolate representing the community-associated MRSA lineage USA400 (Fig. S4C). Interestingly, clade III genomes carried blaZ, tet(L), and aph(3')-III on a similar multi-replicon plasmid (rep5a/rep16), designated pST2250-2, which is approximately 30.3 kb in size (Fig. S4B). Comparative genomics indicated that pST2250-2 shared a conserved backbone with pST2250-1 but contained an additional accessory module carrying tet(L), aph(3')-III, and the heavy metal resistance gene czcD (Fig. S4C), suggesting a history of plasmid recombination. Moreover, pST2250-2 showed the highest sequence similarity (87% coverage and 99.53% identity) to an unnamed plasmid from S. aureus strain Sau41 (CP141473.1), with key differences localized to the resistance gene region, where Sau41’s plasmid carried tet(K) instead of tet(L) (Fig. S4C). Bayesian phylogenetic inference indicated that pST2250-2 was introduced into the ancestor of the MDR subclade of clade III around the year 2001 (Fig. S1) and subsequently underwent multiple gene loss events.
Aside from the fosfomycin resistance gene fosB (98.92%), all other resistance genes were found at low frequencies (<10%) and showed scattered presence across the ST2250 clades.
Comparison of virulence genes in ST2250 clades
The virulence-associated gene content of each clade was characterized across all 277 ST2250 genomes to determine their pathogenic potential. A total of 119 virulence genes were identified, of which 99 (83.19%) were present in more than 90% of genomes across all clades, suggesting that these genes constitute the core virulence gene set of the ST2250 population. As shown in Fig. 3B, the number of virulence genes per genome was significantly higher in clades II (mean = 104.67) and III (mean = 104.61) than in clade I (mean = 103.75), primarily due to the elevated prevalence of the secretion system gene esaG7 in clades II (96.26%) and III (90.67%) compared with clade I (31.25%). In addition, immune evasion cluster (IEC) genes such as scn and sak were found across all clades, with slightly higher detection rates in clades II (75.70%) and III (78.00%) than in clade I (56.25%). Notably, the PVL genes lukF/PV and lukS/PV were detected in 13 ST2250 genomes, while the exfoliative toxin gene eta was identified in one genome (Fig. 3D).
Characterization of the large MGEs in ST2250 lineage
To investigate the potential contribution of large MGEs to genomic plasticity in the ST2250 lineage, we examined the distribution of staphylococcal pathogenicity islands (SaPIs), genomic islands, and prophages across the data set. The overall number of MGEs did not differ significantly among the three clades (P > 0.05, Fig. 3C). As shown in Fig. 3D, the genomic islands vSaα, vSaβ, and vSaγ were found in all genomes analyzed. In contrast, the prevalence of the prophage φSa3 was relatively low across clades I to III (56.25%, 75.70%, and 77.33%, respectively). Notably, although nearly all ST2250 genomes (98.56%) carried SaPI2, only 4.33% (12/277) retained the intact structure; in over 95% of genomes, the conserved region responsible for regulation and replication was missing (Fig. 4A). In addition, one clade II genome and 12 clade III genomes were found to carry φSa2 encoding PVL (Fig. 4B). Of particular interest, one clade II genome (H1864) harbored φSPβ encoding the adhesion gene sasX, which was previously associated with the successful spread of hospital-associated MRSA ST239 in Asia (38).
Comparative genomic analysis of SaPI and prophages in ST2250 genomes. (A, B) Linear comparison of SaPI2 and φSa2 regions between ST2250 genomes and their respective references. (C–F) Structural comparisons of four prophage elements (φST2250-1 to φST2250-4) with their closest known counterparts. Arrowed boxes represent predicted open reading frames, shaded according to functional categories.
We also identified four previously uncharacterized prophages in the ST2250 lineage, designated φST2250-1 through φST2250-4, none of which carried known resistance or virulence genes (Fig. 3D and 4C through F). Structural analysis revealed that all four prophages exhibited a highly organized genomic architecture, including regions associated with lysogeny, DNA replication and transcriptional regulation, packaging, head and tail proteins, and host cell lysis. Notably, sequence differences between φST2250-1 to φST2250-4 and their closest homologs were primarily concentrated in the DNA replication/transcriptional regulation and lysogeny modules, suggesting that these prophages may have undergone recombination during their evolutionary history.
Distributions of defense and anti-defense systems among ST2250 clades
By investigating the defense systems that potentially limit horizontal gene transfer (HGT) and the co-evolved anti-defense mechanisms in the ST2250 lineage, we identified a total of 17 defense systems and 4 anti-defense systems across the data set. In all clades, the number of defense systems per genome was significantly higher than that of anti-defense systems (P < 0.0001, Fig. 5A). Notably, the systems Abi2, AbiP2, type III-A CRISPR-Cas, type I RM, and gcu233 were highly prevalent (>80%) across all clades (Fig. 5B). Given that the type III-A CRISPR-Cas system in both S. epidermidis and S. aureus has been shown to protect against phage infection and plasmid transfer, we further analyzed CRISPR spacers in 270 ST2250 genomes that carried this system. We identified 11 unique spacer sequences, 9 of which shared homology with known phage sequences, and 2 of which targeted S. aureus plasmids (Fig. 5C; Table S2). With the exception of one spacer (IIIASP-11), all other spacers were present in >55% of CRISPR-positive genomes.
*Distributions of defense and anti-defense systems among ST2250 genomes. (A) Quantification of defense and anti-defense systems across the three clades of ST2250. ***, P < 0.0001. (B) Heatmap illustrating the presence or absence of specific defense and anti-defense systems across all ST2250 genomes. (C) Pie chart summarizing the predicted targets of CRISPR spacers among ST2250 genomes.
Open pan-genome architecture of the ST2250 lineage
To investigate the genomic diversity of the ST2250 lineage, we constructed a comprehensive pan-genome comprising core, accessory, and unique genes across all genomes. The resulting ST2250 pangenome consisted of 3,961 genes, with core, accessory, and unique genes accounting for 54.43%, 27.34%, and 18.23%, respectively. A generalized linear model based on the time-calibrated core-genome phylogeny revealed a significant association between core-genome branch length and the number of gene exchange events (P = 2.98e-28), indicating that the ST2250 pan-genome is actively evolving and represents an open genomic structure. Furthermore, pan-genome accumulation curves for each clade continued to rise without reaching a plateau as more genomes were incorporated (Fig. 6A), reinforcing the open nature of the ST2250 pan-genome. This interpretation was further supported by α-values derived from Heap’s law fitting, all of which were below 1 (α = 0.77, 0.84, and 0.85 for clades I, II, and III, respectively). After excluding clade-specific core genes, we found that 16.16% to 31.02% of the variable genes were clade-specific (Fig. 6B and C), indicating moderate interclade variability in the pan-genome.
Pan-genome diversity of ST2250 genomes. (A) Pan-genome accumulation curves for the three clades of ST2250. The total number of gene clusters increases with the number of genomes sampled, and fitted Heaps’ law alpha values are shown for each clade. (B) Proportional distribution of clade-specific vs shared accessory genes across the three clades. Red bars represent genes unique to one clade, while blue bars indicate genes shared with at least one other clade. (C) Binary presence-absence heatmap of accessory genes across all 277 ST2250 genomes, sorted according to the core genome phylogeny. (D) COG functional classification of core, accessory, and unique genes for each of the three clades.
To assess the functional composition of the pan-genome, we performed COG annotation for each clade (Fig. 6D). The functional profiles of core genes were highly similar across clades; in contrast, the accessory and unique genes displayed greater functional diversity across clades. Compared to the core genome, the accessory and unique gene pools of different clades contained a larger number of genes related to functional categories such as “Mobilome: prophages, transposons” and “Defense mechanisms.”
Positive selection analysis of the ST2250 lineage
To further elucidate the evolutionary trajectory of the ST2250 population, we investigated signatures of convergent evolution that may reflect shared adaptive responses to selective pressures. A total of 39 genes were predicted to be under positive selection, with adjusted dN/dS ratios ranging from 1.65 to 2.69 (Table S3; Fig. S5). Among these, two genes were associated with antimicrobial resistance, including fmtB (related to β-lactam resistance) and tcaB (involved in teicoplanin resistance), and two genes were linked to bacterial virulence, including geh (a lipase gene) and kdpD (encoding a sensor histidine kinase implicated in virulence regulation). The remaining 35 genes under positive selection were primarily involved in metabolic processes, particularly in pathways related to amino acid transport and metabolism, carbohydrate transport and metabolism, and energy production and conversion.
Comparison with epidemic lineages of S. aureus
To provide an initial comparison with epidemic lineages of S. aureus, we compared ST2250 with community-associated S. aureus ST59 (prevalent in Asia) and ST8 (prevalent in the United States), and hospital-associated S. aureus ST239 (prevalent in Asia) for AMR genes, virulence genes, and large MGEs (Fig. S6). As shown in Fig. S6A, ST2250 carried a similar number of AMR genes to ST59 (P > 0.05) but significantly fewer than ST8 and ST239 (mean 3.39 vs 5.83 and 12.90; P < 0.001). The higher burden in ST8 and especially ST239 reflected a broader repertoire and higher prevalence of aminoglycoside, macrolide, and tetracycline resistance genes, together with lineage-typical mutations affecting fluoroquinolones, rifampicin, fusidic acid, and trimethoprim, whereas ST2250 uniquely carried the tetracycline resistance gene tet(L). Among the 160 virulence loci identified in the data set, 88 (55%) were present at >90% prevalence in all lineages; nonetheless, ST2250 harbored fewer virulence genes than the S. aureus lineages (Fig. S6B), notably lacking loci across multiple functional categories, including the adhesin gene sasG, the serine protease spl cluster, and a series of enterotoxin genes. Furthermore, the large MGE burden in ST2250 was comparable to ST59 but lower than ST8 and ST239 (mean 5.60 vs 6.70 for ST8; 5.60 vs 7.30 for ST239; P < 0.001; Fig. S6C). Relative to the epidemic S. aureus lineages, ST2250 lacked specific SaPIs, including SaPI2/SaPI3/SaPI4-like elements and SaPI5, the genomic island ACME, and the prophages φSa4 and φSa6, and it showed a lower prevalence of φSa3.
DISCUSSION
Although S. argenteus was recognized as a distinct species around 2015 (4), it was not distinguished from S. aureus, and its global epidemiology remains incompletely understood. In our collection, S. argenteus accounted for 3.91% of presumptive S. aureus isolates, with the ST2250 lineage predominating (82.35%), a prevalence notably higher than that reported previously in East China (0.7%) (39) and Japan (0.55%) (40). Analysis of publicly available ST2250 genome data (n = 277, including 14 isolates from our collection) revealed that ST2250 represents a globally distributed lineage with ongoing transmission and potential environmental or community reservoirs and that its actual epidemiological burden has likely been underestimated due to historical non-differentiation from S. aureus.
Our phylogenomic analysis revealed that the global S. argenteus ST2250 population has diverged into three well-defined clades: clade I branched near the root around 1989, whereas clades II and III shared a recent common ancestor and diversified later, in about 1996 and 1997, respectively, reflecting a long evolutionary history and wide temporal spread. Each clade exhibited distinct geographical clustering, suggesting that ST2250 diversified through region-specific evolutionary events rather than a single global expansion. Phylogenomics together with paired SNP distance analysis revealed long-distance transmission, with clusters spanning Thailand, the United Kingdom, Vietnam, and Denmark, consistent with international spread via frequent human movement or other transmission vectors. Furthermore, non-human ST2250 genomes were phylogenetically intermixed with human-derived genomes, suggesting possible cross-host transmission and the presence of animal or food reservoirs (37). This aligns with a prior report of rapid dissemination of S. argenteus from food handlers to utensils (41) and with an epidemiological study of Chinese retail foods reporting a 7.2% isolation rate of S. argenteus (10), supporting the potential for foodborne or zoonotic transmission. Taken together, these findings point to an ecological network spanning hosts and continents that may facilitate the persistence and global spread of ST2250, underscoring the need for strengthened surveillance across countries and region-specific epidemiological control measures.
Interestingly, our origin analysis supports recent diversification of CC2250 from a deeply split ancestor, rather than recent formation by segmental recombination from CC75. Plausible origin scenarios include a host or ecological shift from an unsampled non-human or environmental reservoir into humans in the mid-1980s with subsequent expansion, or long-term low-level circulation with more recent ecological opportunity or mobile element gains facilitating spread and detection. Discriminating between these scenarios will require broader One Health sampling across animals, foods, environmental and wastewater sources, together with long-read sequencing to resolve structural variants and plasmidomes, metagenomic read mining for CC2250-like sequences, and longitudinal sampling in regions with high prevalence.
The methicillin-resistant phenotype of S. argenteus is relatively rare and exhibits regional variability (19). However, the evolutionary emergence and genomic features of MRSArg remain largely uncharacterized in a global context. Most ST2250 MRSArg in our data set belonged to clade II (94.06%), which formed a tightly clustered sublineage carrying SCCmec IVc, with dfrG (trimethoprim resistance) located within the element. These MRSArg genomes were closely related to MSSAarg genomes within the same clade, suggesting a single SCCmec acquisition event in the early 2000s, analogous to the evolution of other MRSA lineages (42, 43), where SCCmec was acquired once and the clone subsequently radiated.
Additionally, clade II MRSArg genomes universally carried a 20.6 kb multi-replicon plasmid pST2250-1, encoding blaZ and heavy metal resistance genes (cadD, arsR). Similarly, clade III MSSArg genomes formed a multidrug-resistant subclade that exclusively carried a related 30 kb plasmid pST2250-2, derived from pST2250-1, encoding blaZ, tet(L) (tetracycline resistance), aph(3')-III (aminoglycoside resistance), and the heavy metal resistance genes (cadD, arsR, and czcD). Both plasmids were highly similar to their S. aureus counterparts, implying interspecies plasmid transfer between S. aureus and S. argenteus. Importantly, the accumulation of antibiotic and heavy metal resistance determinants likely conferred a selective advantage under hospital-associated pressures, thereby facilitating the spread and persistence of clade II/III genomes in healthcare environments. Furthermore, clades II and III exhibited a higher prevalence of virulence factors compared to clade I, particularly the esaG7 gene in the type VII secretion system, which encodes an antitoxin-like immunity protein that neutralizes the EsaD nuclease toxin, thereby maintaining T7SS stability and enhancing the competitive fitness and persistence of bacteria within host niches (44). In summary, the dominance of clades II and III in global ST2250 S. argenteus populations is likely driven by their expanded resistance and virulence gene profiles, thereby promoting their persistence and transmission.
Beyond SCCmec and plasmids, all ST2250 genomes harbored canonical genomic islands (vSaα/β/γ). Notably, we identified one clade II genome (H1864) that harbored φSPβ, introducing sasX (a critical determinant of MRSA pathogenic success [38]) into a previously unobserved genetic background. This observation underscores the potential for the co-circulating lineages of S. argenteus and S. aureus to exchange bacteriophages and transfer virulence determinants, potentially generating new epidemic variants adapted to competing populations. However, other large MGEs were relatively scarce. For instance, φSa3, which carries IEC genes such as scn and sak, was present in only 56.25%–77.33% of genomes across clades, much lower than its prevalence (>90%) in the S. aureus epidemic lineages (36). Furthermore, the SaPI2 was fragmented in over 95% of genomes, losing conserved regions responsible for regulation and replication. Considering the notably smaller accessory genome in S. argenteus compared to S. aureus (16), we hypothesize that active CRISPR-Cas immunity in ST2250 may limit the uptake of new MGEs. However, although type III-A CRISPR-Cas was highly prevalent in ST2250, we identified only 11 unique spacers within these CRISPR arrays. This low spacer diversity is consistent with shared ancestry of spacer arrays and limited recent spacer acquisition, rather than convergent acquisition across clades, and does not by itself indicate ongoing CRISPR activity. Together with prior work showing that S. argenteus genomes lacking CRISPR-Cas do not harbor more integrated prophages (17), these observations suggest that CRISPR is unlikely to be the sole driver of the MGE landscape in ST2250. We, therefore, interpret the reduced MGE burden as the combined effect of multiple defense systems, including type I RM and abortive infection, along with ecological and temporal factors. In particular, since the inferred origin of ST2250 dates to the mid 1980s, the lineage has had a shorter residence time in the human niche than S. aureus, which may also contribute to its smaller accessory pangenome. Functional assays and longitudinal sampling will be needed to quantify the relative contributions of these mechanisms.
Interestingly, pan-genome analysis revealed that ST2250 lineage has an open genome, with accessory genes continuing to accumulate over time. This pattern may reflect a balance under strong selective pressures: while ST2250 is capable of acquiring beneficial genes, such as plasmid-encoded resistance determinants, the defense systems may limit the integration of random MGEs. In addition, signals of convergent evolution support the idea that ST2250 is adapting to the hospital setting. We identified 39 genes under positive selection across the lineage, spanning functional categories of antibiotic resistance, bacterial virulence, and central metabolism. This distribution suggests that ST2250 faces multiple selective pressures from both antimicrobial exposure and host immune responses, potentially driving selection for traits that enhance both resistance and overall fitness. Similar multigenic adaptations have been observed in hospital-adapted strains of S. aureus and other pathogens subjected to high antibiotic pressure (45, 46). Although experimental validation is still needed, these positively selected mutations likely contribute to the persistence and pathogenicity of ST2250 in nosocomial environments.
This study has several limitations. First, sampling bias is inevitable: nearly half of the global genomes analyzed originate from the Netherlands and Thailand, potentially overrepresenting local diversity and underrepresenting other regions. In addition, public genomes may include copy or serial isolates that cannot be confidently identified across studies, further overrepresenting specific outbreaks or regions. Second, inferred transmission events are based solely on SNP thresholds and phylogenetic proximity, without supporting epidemiological data; thus, transmission directionality and person-to-person spread cannot be confirmed. Third, although the study includes a One Health perspective, non-human genomes are limited (13 out of 277) and represent a narrow range of hosts, which constrains conclusions about cross-species transmission. Future work should broaden geographic and host sampling (including animal and environmental sources) and incorporate functional studies to elucidate the evolutionary dynamics and adaptive mechanisms underlying the global success of ST2250. Additionally, our cross-lineage comparison with S. aureus is preliminary and limited to ST59, ST8, and ST239; a comprehensive and balanced cross-species, multi-lineage analysis that includes additional S. argenteus lineages and a broader set of S. aureus lineages is warranted, together with a formal reconstruction of the ~1986 ancestor, including ancestral core sequence and accessory gene content under explicit gain/loss and HGT models.
In conclusion, this study provides a comprehensive genomic analysis of the globally distributed S. argenteus ST2250 lineage, integrating local clinical isolates with international genome data. We demonstrate that ST2250 has diversified into a basal clade I and sister clades II and III with distinct geographic structures, reflecting region-specific evolutionary events and sustained global spread over the past three decades. Frequent cross-country, intercontinental, and cross-host transmission events were identified, highlighting the ecological versatility of this lineage. Methicillin resistance was almost exclusively confined to clade II, which harbored SCCmec IVc and a blaZ-encoding plasmid, while clade III formed a distinct MDR subclade enriched for other resistance determinants. Both clades II and III also showed increased virulence gene content, suggesting parallel adaptations to hospital-associated environments through different evolutionary strategies. Despite active defense systems that might limit the acquisition of MGEs, the ST2250 pan-genome remains open, with continued gene flux and convergent evolutionary signals targeting resistance and metabolism-related pathways. These findings collectively reveal a dynamic interplay between antimicrobial selection, genomic plasticity, and ecological adaptation underlying the global success of ST2250.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chew KL, Octavia S, Lai D, Lin RTP, Teo JWP. 2021. Staphylococcus singaporensis sp. nov., a new member of the Staphylococcus aureus complex, isolated from human clinical specimens. Int J Syst Evol Microbiol 71. doi:10.1099/ijsem.0.00506734698625 · doi ↗ · pubmed ↗
- 2Schutte AHJ, Strepis N, Zandijk WHA, Bexkens ML, Bode LGM, Klaassen CHW. 2021. Characterization of Staphylococcus roterodami sp. nov., a new species within the Staphylococcus aureus complex isolated from a human foot infection. Int J Syst Evol Microbiol 71. doi:10.1099/ijsem.0.00499634582327 · doi ↗ · pubmed ↗
- 3Mc Donald M, Dougall A, Holt D, Huygens F, Oppedisano F, Giffard PM, Inman-Bamber J, Stephens AJ, Towers R, Carapetis JR, Currie BJ. 2006. Use of a single-nucleotide polymorphism genotyping system to demonstrate the unique epidemiology of methicillin-resistant Staphylococcus aureus in remote aboriginal communities. J Clin Microbiol 44:3720–3727. doi:10.1128/JCM.00836-0617021102 PMC 1594797 · doi ↗ · pubmed ↗
- 4Tong SYC, Schaumburg F, Ellington MJ, Corander J, Pichon B, Leendertz F, Bentley SD, Parkhill J, Holt DC, Peters G, Giffard PM. 2015. Novel staphylococcal species that form part of a Staphylococcus aureus-related complex: the non-pigmented Staphylococcus argenteus sp. nov. and the non-human primate-associated Staphylococcus schweitzeri sp. nov. Int J Syst Evol Microbiol 65:15–22. doi:10.1099/ijs.0.062752-025269845 PMC 4298100 · doi ↗ · pubmed ↗
- 5Thaipadungpanit J, Amornchai P, Nickerson EK, Wongsuvan G, Wuthiekanun V, Limmathurotsakul D, Peacock SJ. 2015. Clinical and molecular epidemiology of Staphylococcus argenteus infections in Thailand. J Clin Microbiol 53:1005–1008. doi:10.1128/JCM.03049-1425568440 PMC 4390622 · doi ↗ · pubmed ↗
- 6Kitagawa H, Ohge H, Hisatsune J, Masuda K, Aziz F, Hara T, Kuroo Y, Sugai M. 2020. Low incidence of Staphylococcus argenteus bacteremia in Hiroshima, Japan. J Infect Chemother 26:140–143. doi:10.1016/j.jiac.2019.07.01131377128 · doi ↗ · pubmed ↗
- 7Jauneikaite E, Pichon B, Mosavie M, Fallowfield JL, Davey T, Thorpe N, Nelstrop A, Sriskandan S, Lamb LE. 2021. Staphylococcus argenteus transmission among healthy Royal Marines: a molecular epidemiology case-study. J Infect 83:550–553. doi:10.1016/j.jinf.2021.08.04034469709 · doi ↗ · pubmed ↗
- 8Rocha Balzan LDL, Rossato AM, Riche CVW, Cantarelli VV, D’Azevedo PA, Valério de Lima A, Rodrigues B, França E, Dias CAG, Sampaio JLM. 2023. Staphylococcus argenteus infections, Brazil. Microbiol Spectr 11:e 0117922. doi:10.1128/spectrum.01179-2236688721 PMC 9927369 · doi ↗ · pubmed ↗